
Prevention of CD8 T Cell Deletion during Chronic Viral Infection

 ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice and Virus
2.2. Flow Cytometry
2.3. Ribavirin Treatment
2.4. Costimulation Blockade
2.5. In Vitro and In Vivo Cytotoxicity Assays
2.6. Statistical Analysis
3. Results
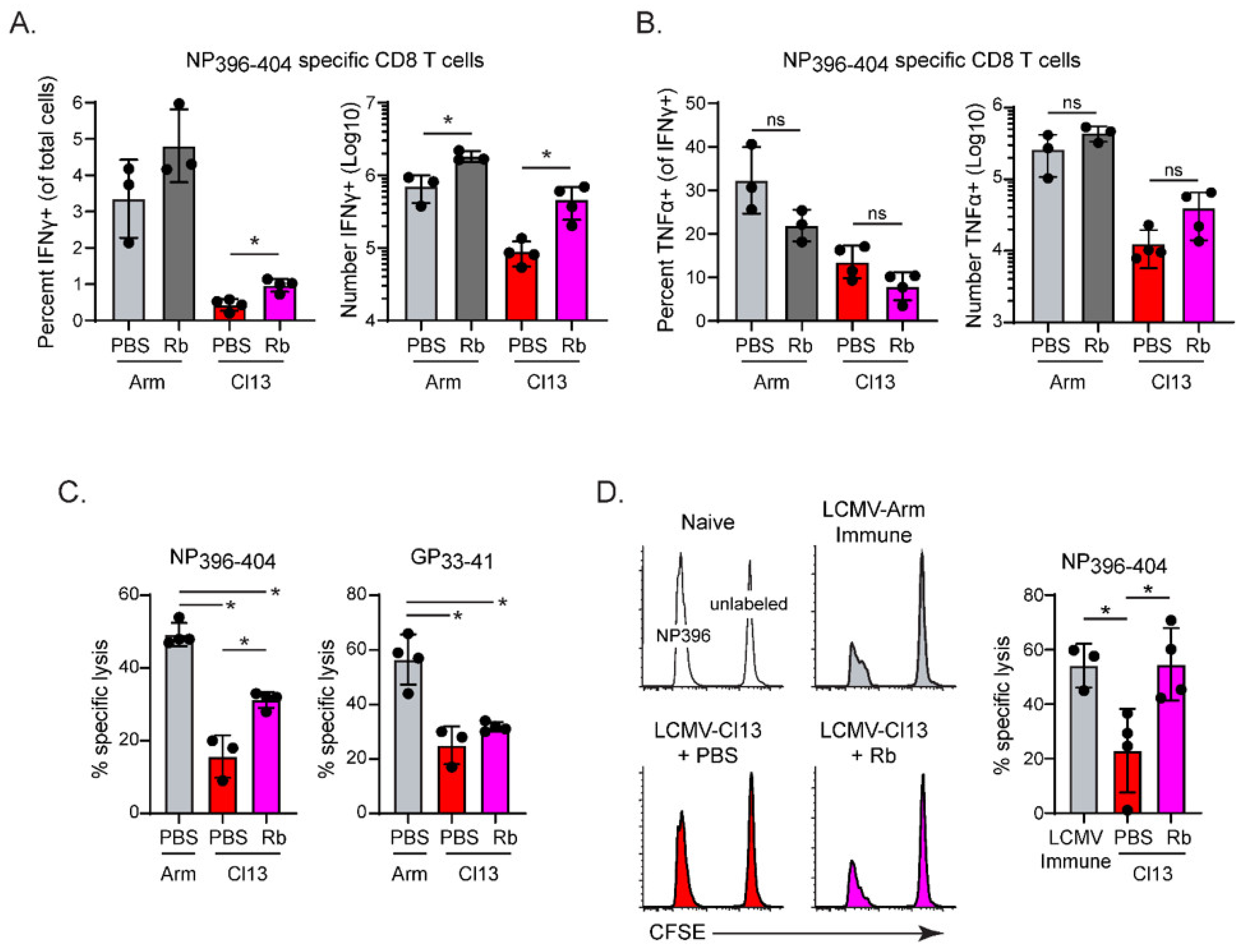
3.1. Lowering Virus Titers during Early Chronic Infection Preserves High-Affinity LCMV-NP396–404 Dpecific CD8 T Cells
3.2. CD4 T Cells Are Required to Preserve High-Affinity LCMV-NP396–404 Specific CD8 T Cells
3.3. Prolonged Costimulatory Interactions Are Required to Sustain Virus-Specific CD4 T Cells and High-Affinity LCMV-NP396–404 Specific CD8 T Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Brooks, D.G.; Trifilo, M.J.; Edelmann, K.H.; Teyton, L.; McGavern, D.B.; Oldstone, M.B. Interleukin-10 determines viral clearance or persistence in vivo. Nat. Med. 2006, 12, 1301–1309. [Google Scholar] [CrossRef]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef] [Green Version]
- Snell, L.M.; McGaha, T.L.; Brooks, D.G. Type I Interferon in Chronic Virus Infection and Cancer. Trends Immunol. 2017, 38, 542–557. [Google Scholar] [CrossRef] [PubMed]
- Zajac, A.J.; Blattman, J.N.; Murali-Krishna, K.; Sourdive, D.J.; Suresh, M.; Altman, J.D.; Ahmed, R. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 1998, 188, 2205–2213. [Google Scholar] [CrossRef] [PubMed]
- Gallimore, A.; Glithero, A.; Godkin, A.; Tissot, A.C.; Pluckthun, A.; Elliott, T.; Hengartner, H.; Zinkernagel, R. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I-peptide complexes. J. Exp. Med. 1998, 187, 1383–1393. [Google Scholar] [CrossRef]
- Oxenius, A.; Price, D.A.; Easterbrook, P.J.; O’Callaghan, C.A.; Kelleher, A.D.; Whelan, J.A.; Sontag, G.; Sewell, A.K.; Phillips, R.E. Early highly active antiretroviral therapy for acute HIV-1 infection preserves immune function of CD8+ and CD4+ T lymphocytes. Proc. Natl. Acad. Sci. USA 2000, 97, 3382–3387. [Google Scholar] [CrossRef] [Green Version]
- Brooks, D.G.; McGavern, D.B.; Oldstone, M.B. Reprogramming of antiviral T cells prevents inactivation and restores T cell activity during persistent viral infection. J. Clin. Investig. 2006, 116, 1675–1685. [Google Scholar] [CrossRef] [Green Version]
- Wherry, E.J.; Blattman, J.N.; Murali-Krishna, K.; van der Most, R.; Ahmed, R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 2003, 77, 4911–4927. [Google Scholar] [CrossRef] [Green Version]
- Matloubian, M.; Concepcion, R.J.; Ahmed, R. CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J. Virol. 1994, 68, 8056–8063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battegay, M.; Moskophidis, D.; Rahemtulla, A.; Hengartner, H.; Mak, T.W.; Zinkernagel, R.M. Enhanced establishment of a virus carrier state in adult CD4+ T-cell-deficient mice. J. Virol. 1994, 68, 4700–4704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubert, R.D.; Kamphorst, A.O.; Sarkar, S.; Vezys, V.; Ha, S.J.; Barber, D.L.; Ye, L.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Antigen-specific CD4 T-cell help rescues exhausted CD8 T cells during chronic viral infection. Proc. Natl. Acad. Sci. USA 2011, 108, 21182–21187. [Google Scholar] [CrossRef] [Green Version]
- Snell, L.M.; Osokine, I.; Yamada, D.H.; De la Fuente, J.R.; Elsaesser, H.J.; Brooks, D.G. Overcoming CD4 Th1 Cell Fate Restrictions to Sustain Antiviral CD8 T Cells and Control Persistent Virus Infection. Cell Rep. 2016, 16, 3286–3296. [Google Scholar] [CrossRef] [Green Version]
- Brooks, D.G.; Teyton, L.; Oldstone, M.B.; McGavern, D.B. Intrinsic functional dysregulation of CD4 T cells occurs rapidly following persistent viral infection. J. Virol. 2005, 79, 10514–10527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, R.; Salmi, A.; Butler, L.D.; Chiller, J.M.; Oldstone, M.B. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J. Exp. Med. 1984, 160, 521–540. [Google Scholar] [CrossRef] [PubMed]
- Oldstone, M.B.A. Virus, Plagues, and History, 2nd ed.; Oxford University Press: Oxford, UK, 2020. [Google Scholar]
- Sakabe, S.; Hartnett, J.N.; Ngo, N.; Goba, A.; Momoh, M.; Sandi, J.D.; Kanneh, L.; Cubitt, B.; Garcia, S.D.; Ware, B.C.; et al. Identification of Common CD8(+) T Cell Epitopes from Lassa Fever Survivors in Nigeria and Sierra Leone. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, B.M.; Sakabe, S.; Hartnett, J.N.; Ngo, N.; Goba, A.; Momoh, M.; Demby Sandi, J.; Kanneh, L.; Cubitt, B.; Garcia, S.D.; et al. High crossreactivity of human T cell responses between Lassa virus lineages. PLoS Pathog. 2020, 16, e1008352. [Google Scholar] [CrossRef]
- Elsaesser, H.; Sauer, K.; Brooks, D.G. IL-21 is required to control chronic viral infection. Science 2009, 324, 1569–1572. [Google Scholar] [CrossRef] [Green Version]
- Yi, J.S.; Du, M.; Zajac, A.J. A vital role for interleukin-21 in the control of a chronic viral infection. Science 2009, 324, 1572–1576. [Google Scholar] [CrossRef]
- Frohlich, A.; Kisielow, J.; Schmitz, I.; Freigang, S.; Shamshiev, A.T.; Weber, J.; Marsland, B.J.; Oxenius, A.; Kopf, M. IL-21R on T cells is critical for sustained functionality and control of chronic viral infection. Science 2009, 324, 1576–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brooks, D.G.; Tishon, A.; Oldstone, M.B.A.; McGavern, D.B. Prevention of CD8 T Cell Deletion during Chronic Viral Infection. Viruses 2021, 13, 1189. https://doi.org/10.3390/v13071189
Brooks DG, Tishon A, Oldstone MBA, McGavern DB. Prevention of CD8 T Cell Deletion during Chronic Viral Infection. Viruses. 2021; 13(7):1189. https://doi.org/10.3390/v13071189
Chicago/Turabian StyleBrooks, David G., Antoinette Tishon, Michael B. A. Oldstone, and Dorian B. McGavern. 2021. "Prevention of CD8 T Cell Deletion during Chronic Viral Infection" Viruses 13, no. 7: 1189. https://doi.org/10.3390/v13071189
APA StyleBrooks, D. G., Tishon, A., Oldstone, M. B. A., & McGavern, D. B. (2021). Prevention of CD8 T Cell Deletion during Chronic Viral Infection. Viruses, 13(7), 1189. https://doi.org/10.3390/v13071189