
A Fluorescent Real-Time Plaque Assay Enables Single-Cell Analysis of Virus-Induced Cytopathic Effect by Live-Cell Imaging



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Viruses
2.2. Live-Cell Imaging-Based Fluorescent Real-Time Plaque Assay
2.3. Live-Cell Imaging-Based Fluorescent Real-Time Plaque Reduction Assay
2.4. Statistics
3. Results
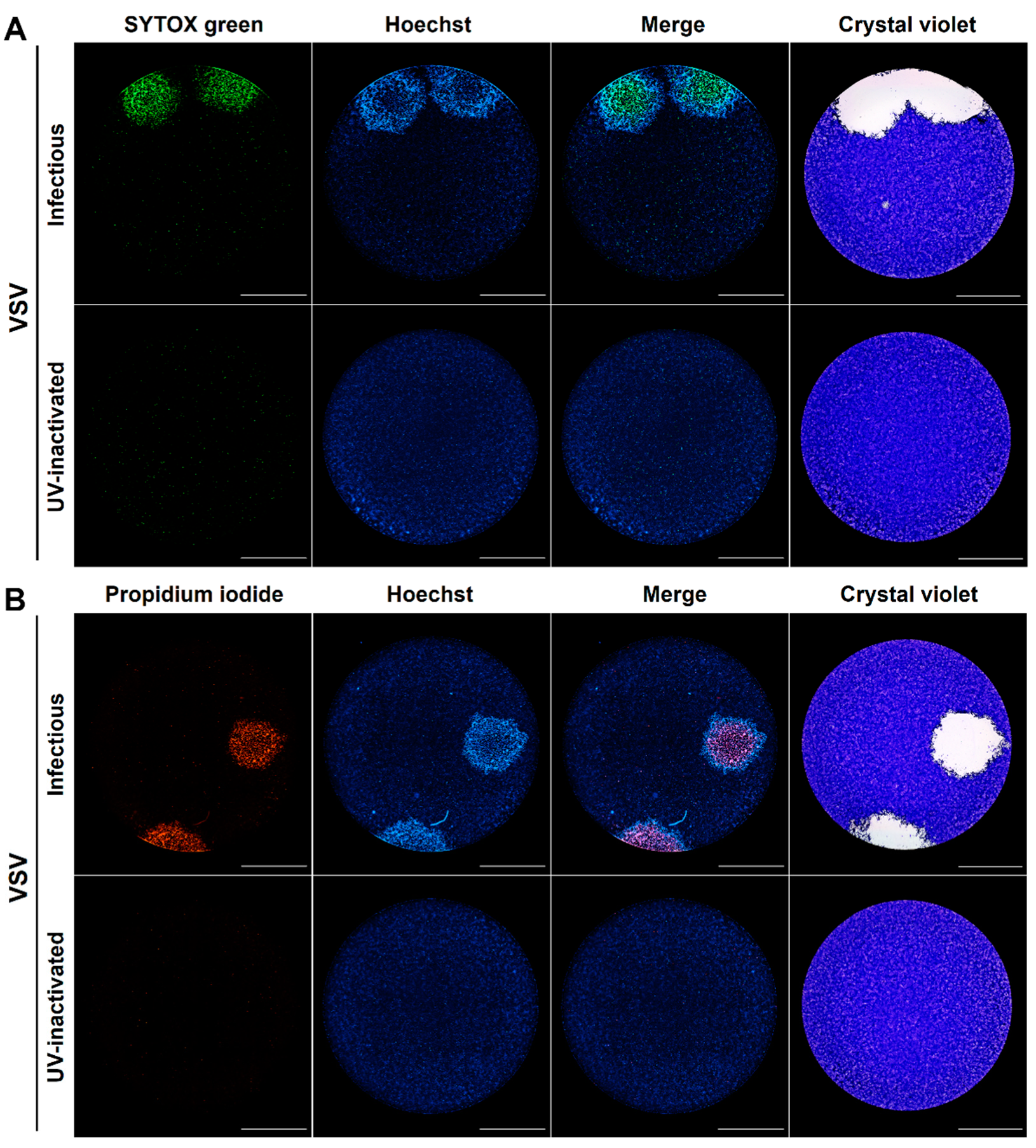
3.1. The Differential Cell Permeability of DNA Fluorescent Dyes Enables the Visualization of Different Stages of the Cytopathic Effect at a Single-Cell Level within Individual Viral Plaques
3.2. Time-Lapse Microscopy of Viral Plaques Labeled with DNA Fluorescent Dyes Enables the Real-Time Kinetic Identification of an Early Chromatin Condensation Wave Followed by a Membrane Permeabilization Wave with Single-Cell Resolution
3.3. The Real-Time Plaque Assay with DNA Stains of Differential Membrane Permeability Enables Automated Identification of Single Viral Plaques with Higher Resolution When Compared to the Standard Crystal Violet Staining
3.4. The Combination of the Real-Time Plaque Assay with Automated Image Analysis Enables a High-Resolution Real-Time Plaque Reduction Assay for the Simultaneous Screening of Drugs in Terms of Antiviral and Cytotoxic Effect
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wen, Z.; Citron, M.; Bett, A.J.; Espeseth, A.S.; Vora, K.A.; Zhang, L.; DiStefano, D.J. Development and application of a higher throughput RSV plaque assay by immunofluorescent imaging. J. Virol. Methods 2019, 263, 88–95. [Google Scholar] [CrossRef]
- d’Hérelle, F. The bacteriophage and its behavior. Nature 1926, 118, 183–185. [Google Scholar] [CrossRef]
- Dulbecco, R. Production of plaques in monolayer tissue cultures by single particles of an animal virus. Proc. Natl. Acad. Sci. USA 1952, 38, 747–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dulbecco, R.; Vogt, M. Plaque formation and isolation of pure lines with poliomyelitis viruses. J. Exp. Med. 1954, 99, 167–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baer, A.; Kehn-Hall, K. Viral concentration determination through plaque assays: Using traditional and novel overlay systems. J. Vis. Exp. 2014, 1–10. [Google Scholar] [CrossRef]
- Mendoza, E.J.; Manguiat, K.; Wood, H.; Drebot, M. Two detailed plaque assay protocols for the quantification of infectious SARS-CoV-2. Curr. Protoc. Microbiol. 2020, 57, cpmc105. [Google Scholar] [CrossRef] [PubMed]
- Masci, A.L.; Menesale, E.B.; Chen, W.C.; Co, C.; Lu, X.; Bergelson, S. Integration of fluorescence detection and image-based automated counting increases speed, sensitivity, and robustness of plaque assays. Mol. Ther. Methods Clin. Dev. 2019, 14, 270–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amarilla, A.A.; Modhiran, N.; Setoh, Y.X.; Peng, N.Y.G.; Sng, J.D.J.; Liang, B.; McMillan, C.L.D.; Freney, M.E.; Cheung, S.T.M.; Chappell, K.J.; et al. An optimized high-throughput immuno-plaque assay for SARS-CoV-2. Front. Microbiol. 2021, 12, 1–17. [Google Scholar] [CrossRef]
- Yakimovich, A.; Andriasyan, V.; Witte, R.; Wang, I.H.; Prasad, V.; Suomalainen, M.; Greber, U.F. Plaque2.0-a high-throughput analysis framework to score virus-cell transmission and clonal cell expansion. PLoS ONE 2015, 10, e0138760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arias-Arias, J.L.; MacPherson, D.J.; Hill, M.E.; Hardy, J.A.; Mora-Rodríguez, R. A fluorescence-activatable reporter of flavivirus NS2B–NS3 protease activity enables live imaging of infection in single cells and viral plaques. J. Biol. Chem. 2020, 295, 2212–2226. [Google Scholar] [CrossRef]
- Farnsworth, A.; Goldsmith, K.; Johnson, D.C. Herpes Simplex virus glycoproteins gD and gE/gI serve essential but redundant functions during acquisition of the virion envelope in the cytoplasm. J. Virol. 2003, 77, 8481–8494. [Google Scholar] [CrossRef] [Green Version]
- Van Remmerden, Y.; Xu, F.; Van Eldik, M.; Heldens, J.G.M.; Huisman, W.; Widjojoatmodjo, M.N. An improved respiratory syncytial virus neutralization assay based on the detection of green fluorescent protein expression and automated plaque counting. Virol. J. 2012, 9, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Tamura, T.; Fukuhara, T.; Uchida, T.; Ono, C.; Mori, H.; Sato, A.; Fauzyah, Y.; Okamoto, T.; Kurosu, T.; Setoh, Y.X.; et al. Characterization of recombinant Flaviviridae viruses possessing a small reporter tag. J. Virol. 2017, 92, e01582-17. [Google Scholar] [CrossRef] [Green Version]
- Arias-Arias, J.L.; Mora-Rodríguez, R. Generation and Implementation of Reporter BHK-21 Cells for Live Imaging of Flavivirus Infection. Bio-Protocol 2021, 11, e3942. [Google Scholar] [CrossRef]
- Xiang, Y.; Cox, H.; Lebedeva, I.; Coleman, J.; Shen, D.; Pande, P.; Schultz, J.; Patton, W.F. A cell-permeant dye for cell cycle analysis by flow and laser-scanning microplate cytometry. Nat. Methods 2009, 6, an2–an3. [Google Scholar] [CrossRef]
- Hubbard, K.S.; Gut, I.M.; Scheeler, S.M.; Lyman, M.E.; McNutt, P.M. Compatibility of SYTO 13 and Hoechst 33342 for longitudinal imaging of neuron viability and cell death. BMC Res. Notes 2012, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wlodkowic, D.; Faley, S.; Darzynkiewicz, Z.; Cooper, J.M. Real-time cytotoxicity assays. Methods Mol. Biol. 2011, 731, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Bishnoi, S.; Tiwari, R.; Gupta, S.; Byrareddy, S.N.; Nayak, D. Oncotargeting by Vesicular Stomatitis Virus (VSV): Advances in cancer therapy. Viruses 2018, 10, 90. [Google Scholar] [CrossRef] [Green Version]
- Yoon, M.; Spear, P.G. Disruption of adherens junctions liberates Nectin-1 to serve as receptor for herpes simplex virus and pseudorabies virus entry. J. Virol. 2002, 76, 7203–7208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh Roy, S.; Sadigh, B.; Datan, E.; Lockshin, R.A.; Zakeri, Z. Regulation of cell survival and death during Flavivirus infections. World J. Biol. Chem. 2014, 5, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Liprandi, F. Isolation of plaque variants differing in virulence from the 17D strain of yellow fever virus. J. Gen. Virol. 1981, 56, 363–370. [Google Scholar] [CrossRef]
- Burckhardt, C.J.; Greber, U.F. Virus movements on the plasma membrane support infection and transmission between cells. PLoS Pathog. 2009, 5, e1000621. [Google Scholar] [CrossRef]
- Mothes, W.; Sherer, N.M.; Jin, J.; Zhong, P. Virus cell-to-cell transmission. J. Virol. 2010, 84, 8360–8368. [Google Scholar] [CrossRef] [Green Version]
- Gadaleta, P.; Vacotto, M.; Coulombié, F. Vesicular stomatitis virus induces apoptosis at early stages in the viral cycle and does not depend on virus replication. Virus Res. 2002, 86, 87–92. [Google Scholar] [CrossRef]
- Quaresma, J.A.S.; Barros, V.L.R.S.; Pagliari, C.; Fernandes, E.R.; Guedes, F.; Takakura, C.F.H.; Andrade, H.F.; Vasconcelos, P.F.C.; Duarte, M.I.S. Revisiting the liver in human yellow fever: Virus-induced apoptosis in hepatocytes associated with TGF-β, TNF-α and NK cells activity. Virology 2006, 345, 22–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Cheng, A.; Wang, M.; Yin, Z.; Jia, R. The dual regulation of apoptosis by flavivirus. Front. Microbiol. 2021, 12, 574. [Google Scholar] [CrossRef] [PubMed]
- Holanda, G.M.; Casseb, S.M.M.; Quaresma, J.A.S.; Vasconcelos, P.F.C.; Cruz, A.C.R. Yellow fever virus modulates cytokine mRNA expression and induces activation of caspase 3/7 in the human hepatocarcinoma cell line HepG2. Arch. Virol. 2019, 164, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Airo, A.M.; Urbanowski, M.D.; Lopez-Orozco, J.; You, J.H.; Skene-Arnold, T.D.; Holmes, C.; Yamshchikov, V.; Malik-Soni, N.; Frappier, L.; Hobman, T.C. Expression of flavivirus capsids enhance the cellular environment for viral replication by activating Akt-signalling pathways. Virology 2018, 516, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; He, S. The interplay between human herpes simplex virus infection and the apoptosis and necroptosis cell death pathways. Virol. J. 2016, 13, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Schepis, A.; Huang, H.; Yang, J.; Ma, W.; Torra, J.; Zhang, S.Q.; Yang, L.; Wu, H.; Nonell, S.; et al. Designing a green fluorogenic protease reporter by flipping a beta strand of GFP for imaging apoptosis in animals. J. Am. Chem. Soc. 2019, 141, 4526–4530. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Uchiyama, Y. A super-ecliptic, phluorin-mkate2, tandem fluorescent protein-tagged human LC3 for the monitoring of mammalian autophagy. PLoS ONE 2014, 9, e110600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaeffer, H.J.; Beauchamp, L.; De Miranda, P.; Elion, G.B.; Bauer, D.J.; Collins, P. 9-(2-Hydroxyethoxymethyl)guanine activity against viruses of the herpes group. Nature 1978, 272, 583–585. [Google Scholar] [CrossRef] [PubMed]
- Crumpacker, C.S.; Schnipper, L.E.; Zaia, J.A.; Levin, M.J. Growth inhibition by acycloguanosine of herpesviruses isolated from human infections. Antimicrob. Agents Chemother. 1979, 15, 642–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrei, G.; Snoeck, R.; Desmyter, J.; Hospital, A. Comparative activity of various compounds against clinical strains of Herpes Simplex Virus. Eur. J. Clin. Microbiol. Infect. Dis. Vol. 1992, 11, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, A.; Bate, B.J.; Masters, H.B.; Schneider, S.A.; Clark, J.C.; Wren, C.G.; Allaman, J.A.; Levin, M.J. In vitro activities of penciclovir and acyclovir against herpes simplex virus types 1 and 2. Antimicrob. Agents Chemother. 1992, 36, 2037–2038. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arias-Arias, J.L.; Corrales-Aguilar, E.; Mora-Rodríguez, R.A. A Fluorescent Real-Time Plaque Assay Enables Single-Cell Analysis of Virus-Induced Cytopathic Effect by Live-Cell Imaging. Viruses 2021, 13, 1193. https://doi.org/10.3390/v13071193
Arias-Arias JL, Corrales-Aguilar E, Mora-Rodríguez RA. A Fluorescent Real-Time Plaque Assay Enables Single-Cell Analysis of Virus-Induced Cytopathic Effect by Live-Cell Imaging. Viruses. 2021; 13(7):1193. https://doi.org/10.3390/v13071193
Chicago/Turabian StyleArias-Arias, Jorge L., Eugenia Corrales-Aguilar, and Rodrigo A. Mora-Rodríguez. 2021. "A Fluorescent Real-Time Plaque Assay Enables Single-Cell Analysis of Virus-Induced Cytopathic Effect by Live-Cell Imaging" Viruses 13, no. 7: 1193. https://doi.org/10.3390/v13071193
APA StyleArias-Arias, J. L., Corrales-Aguilar, E., & Mora-Rodríguez, R. A. (2021). A Fluorescent Real-Time Plaque Assay Enables Single-Cell Analysis of Virus-Induced Cytopathic Effect by Live-Cell Imaging. Viruses, 13(7), 1193. https://doi.org/10.3390/v13071193