
The Erns Carboxyterminus: Much More Than a Membrane Anchor

 , ,
, ,
Abstract
:1. Introduction
2. Material and Methods
2.1. Cells and Viruses
2.2. Construction of Recombinant Plasmids
2.3. Transient Expression, Immunoprecipitation and Quantification of Proteins
2.4. Analysis of Membrane Association
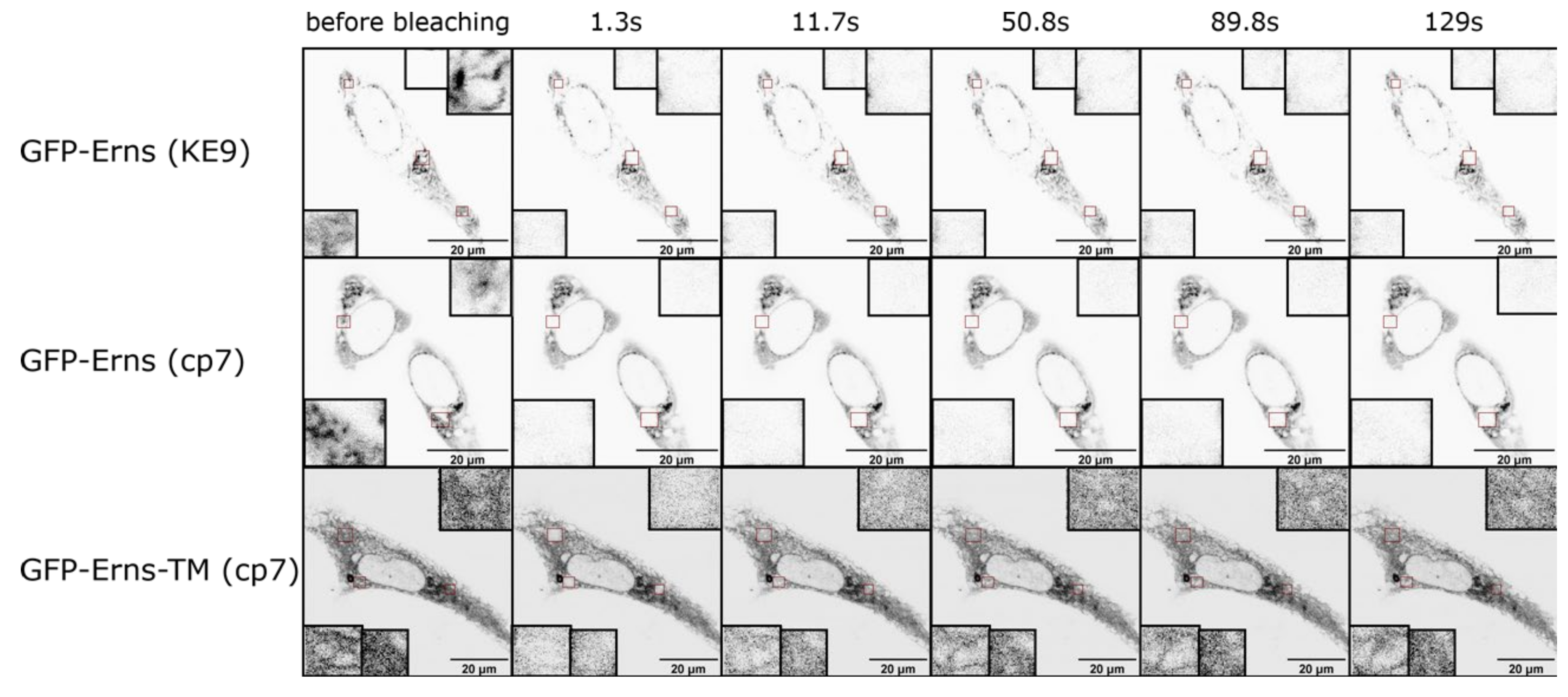
2.5. Fluorescence Recovery after Photobleaching (FRAP) Experiments
2.6. Recovery and Analysis of Mutant Viruses from Cloned Sequences
2.7. Isolation and Characterization of Extracellular Vesicles
2.8. Quantification of Viral Intra- and Extracellular RNA with qRT-PCR
3. Results
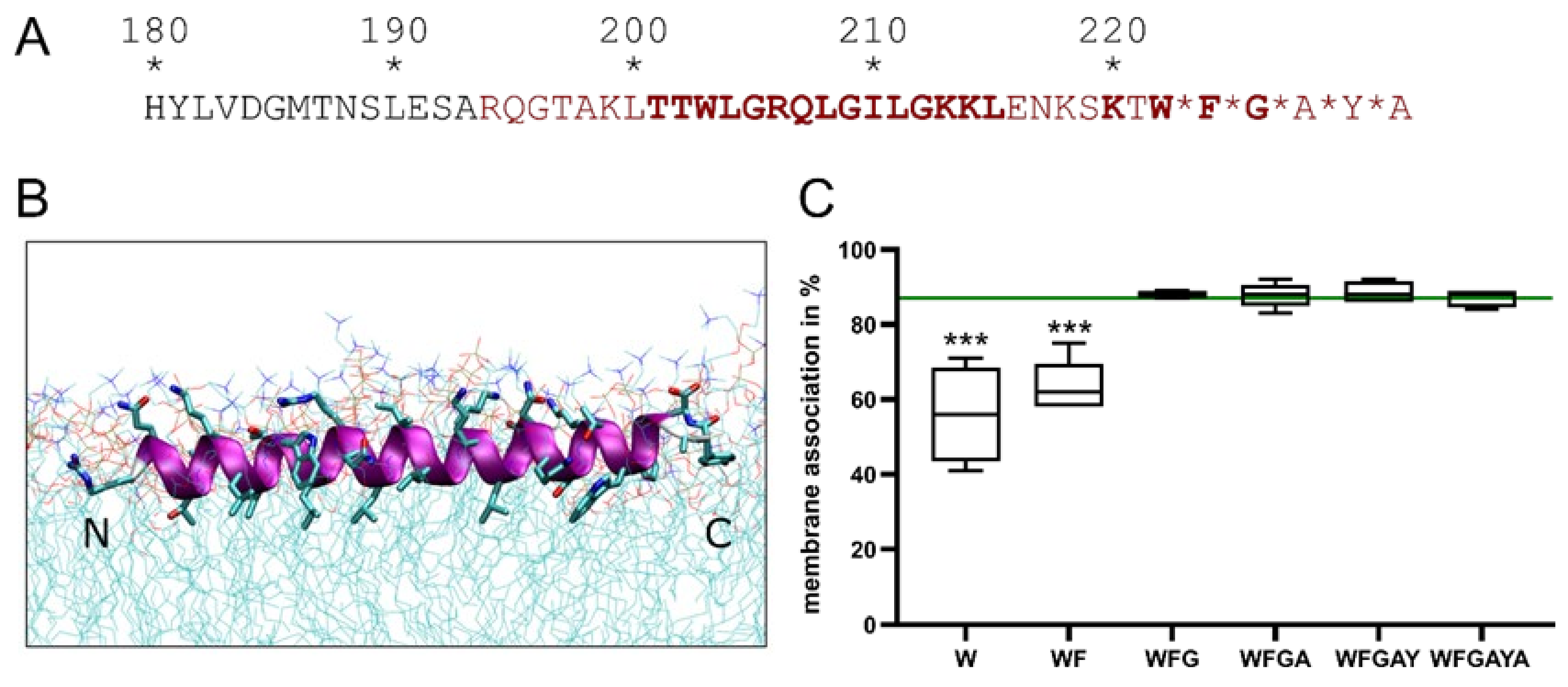
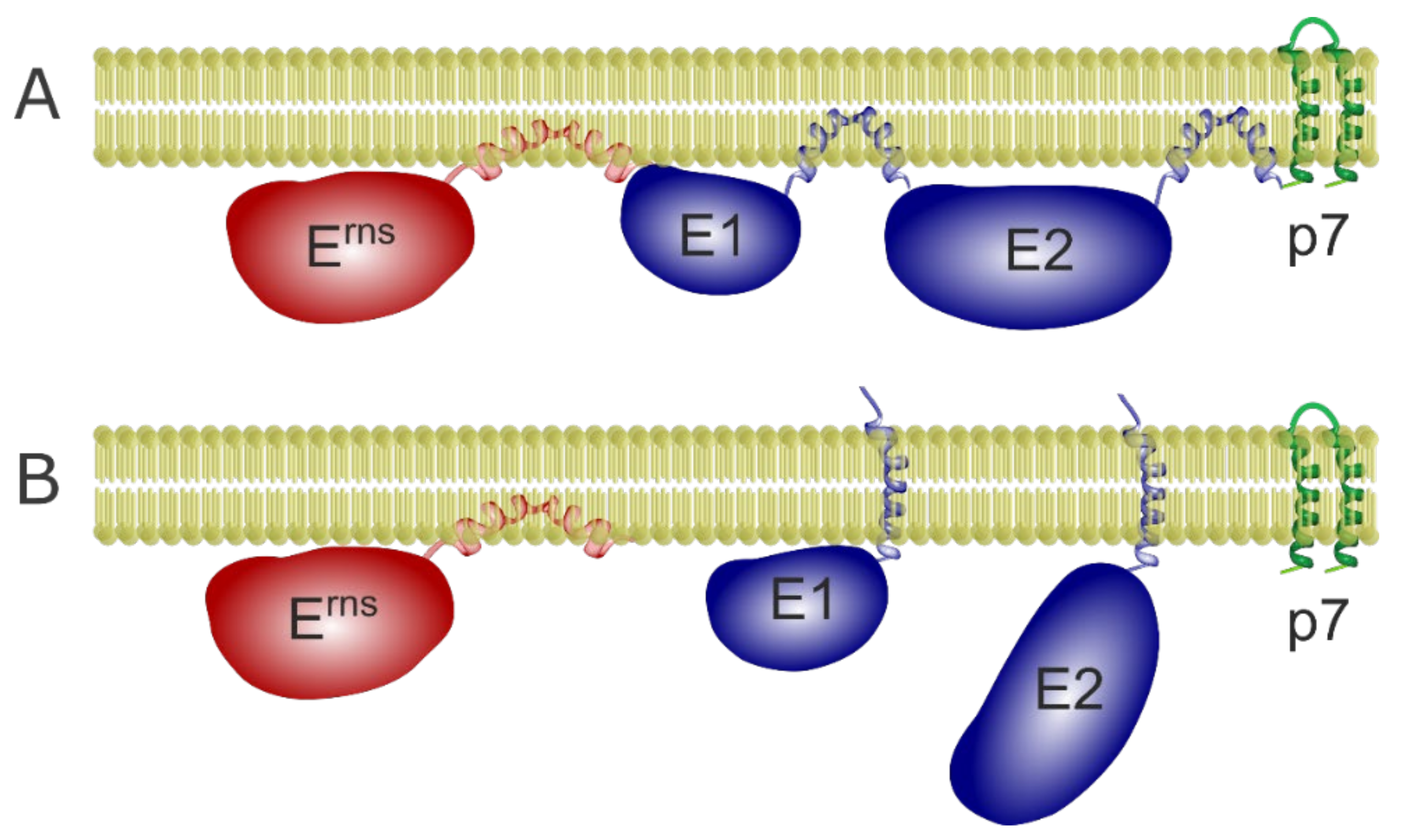
3.1. The Erns Membrane Anchor
3.2. Erns-E1 Processing
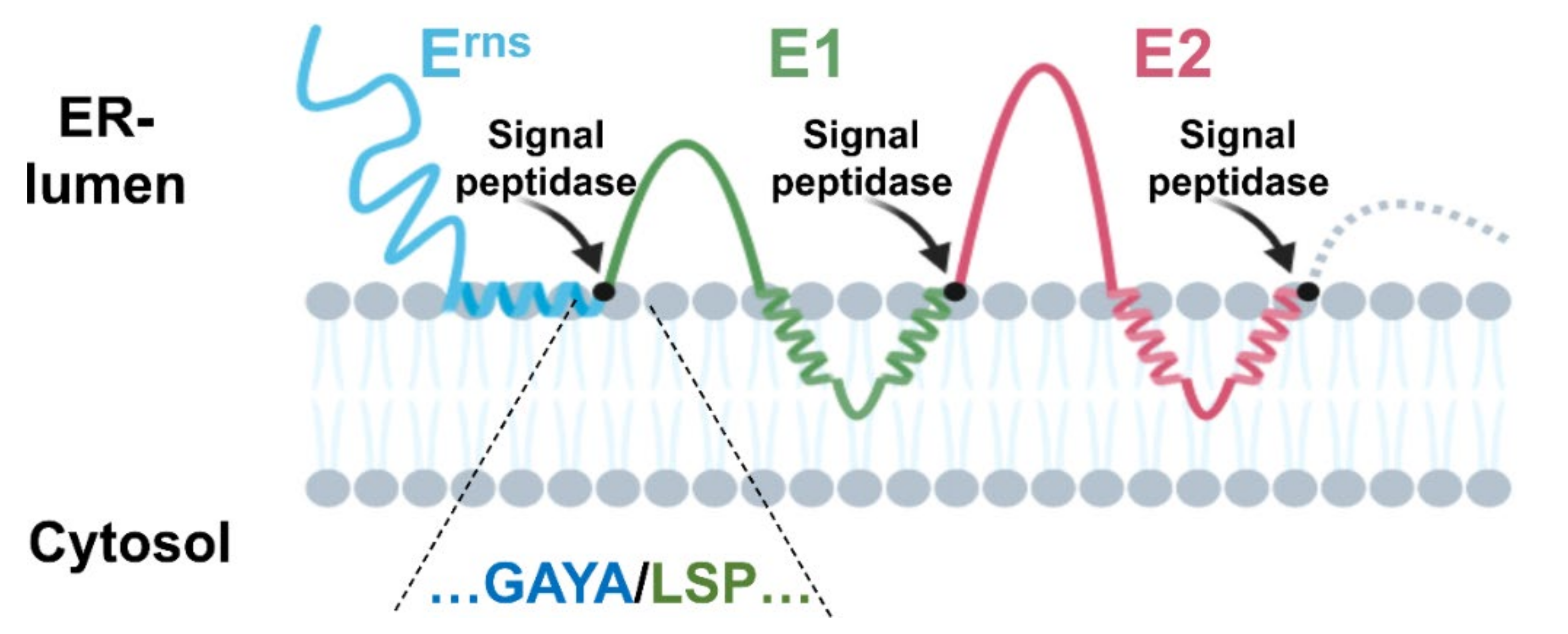
3.2.1. The Erns/E1 Site Is Cleaved by Signal Peptidase

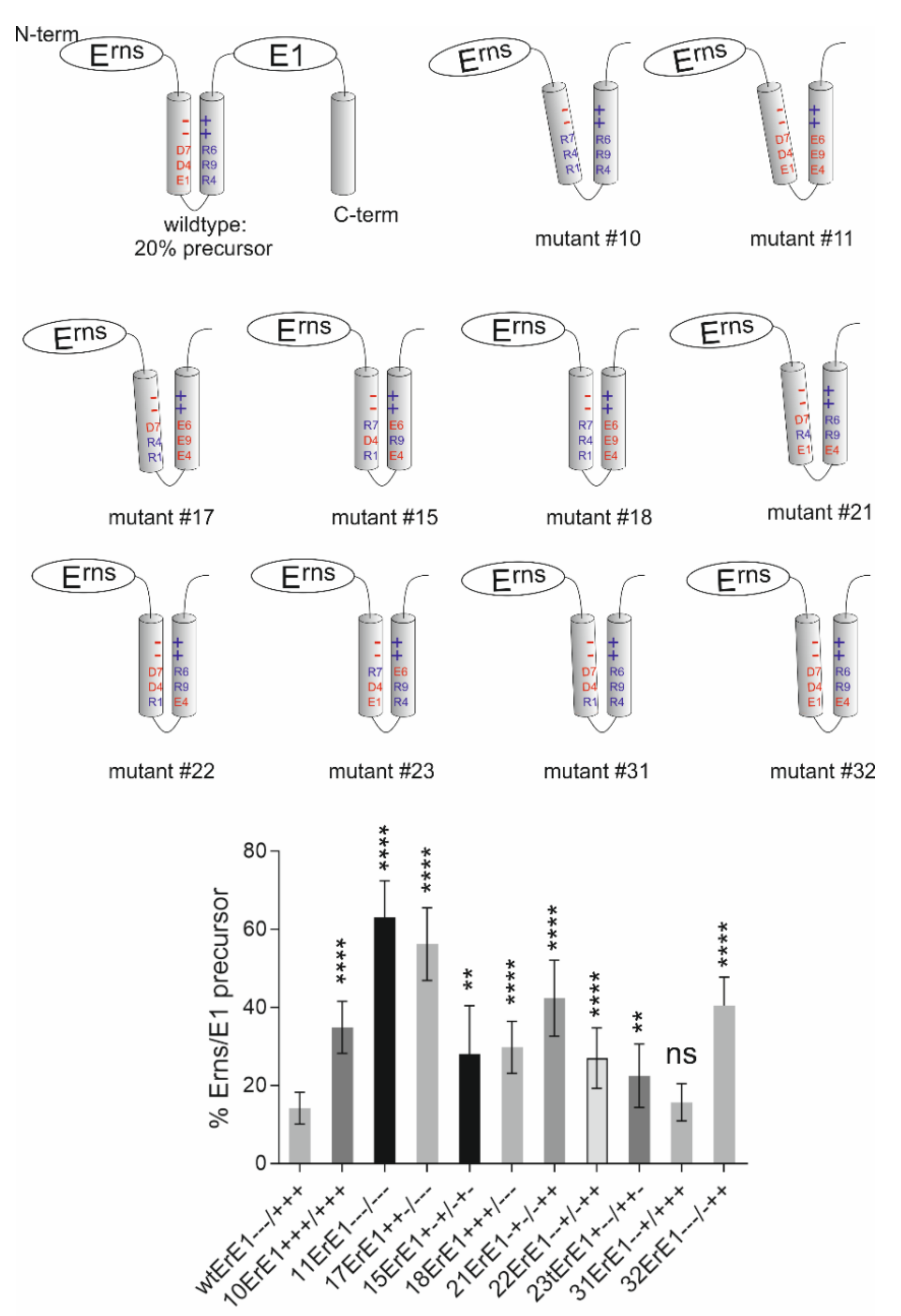
3.2.2. Importance of Specific Amphipathic Helix Residues for Erns-E1 Processing
3.2.3. The Role of E1 and E2 in Erns-E1 Cleavage
3.3. Intracellular Retention and Localization of Erns
3.4. Dimerization of Erns
3.5. E2 as a Further Player
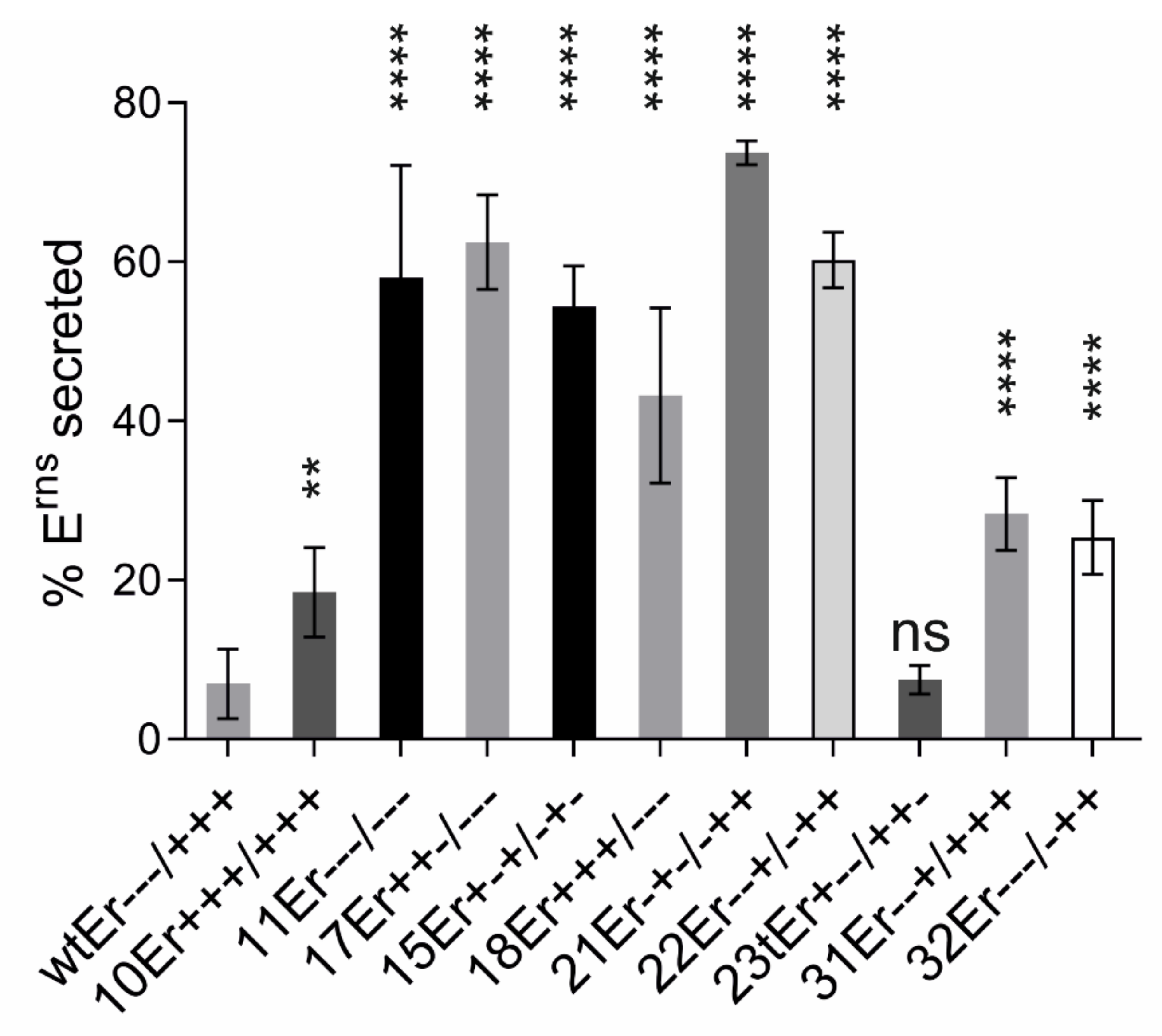
3.6. Secretion of Erns
3.6.1. Importance of Specific Amphipathic Helix Residues for Secretion of Erns
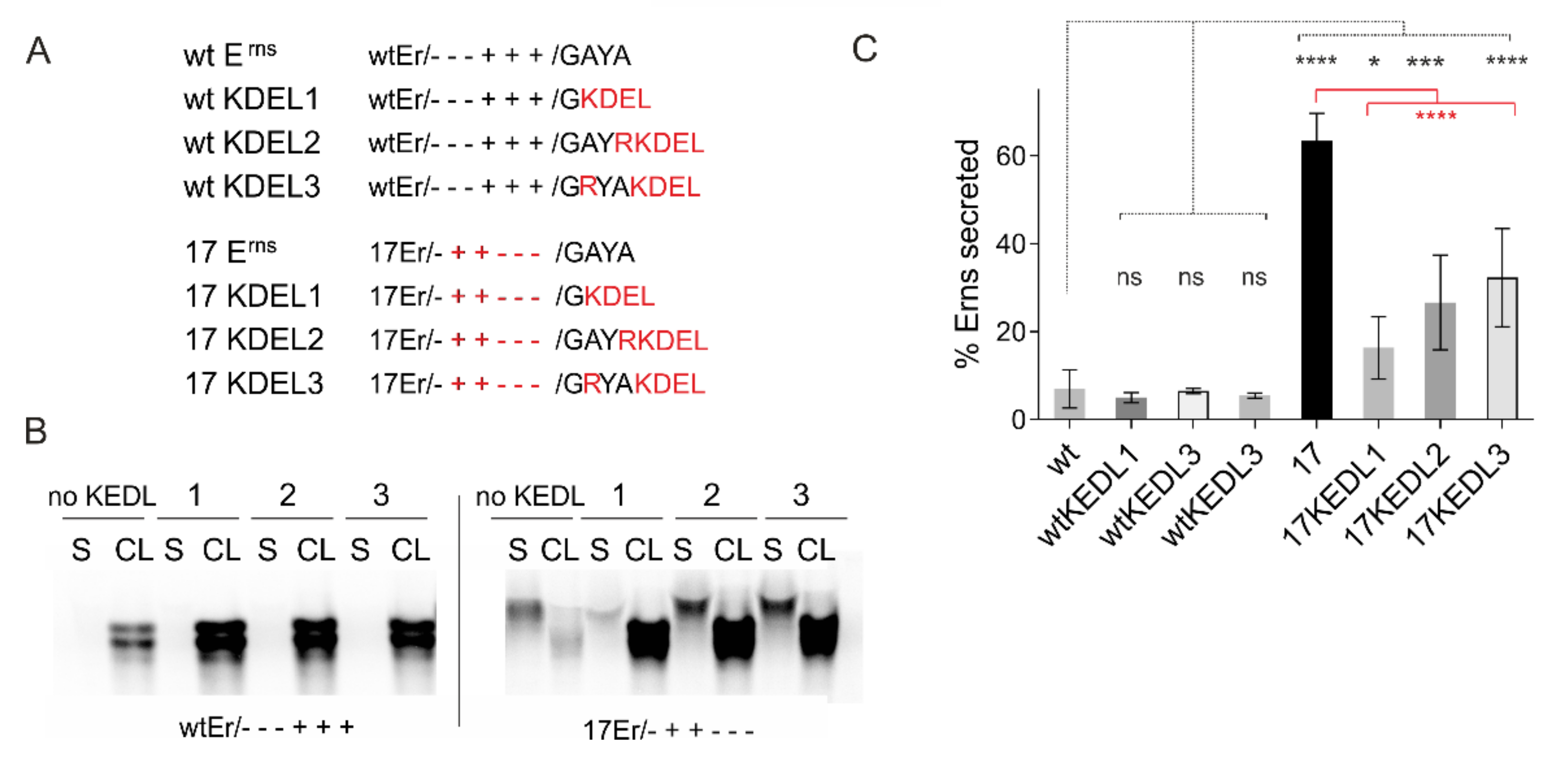
3.6.2. The Role of Carboxy-Terminal Extensions for Secretion of Erns
3.6.3. Not All Erns Synthesized in Cells Is Secreted Over Time
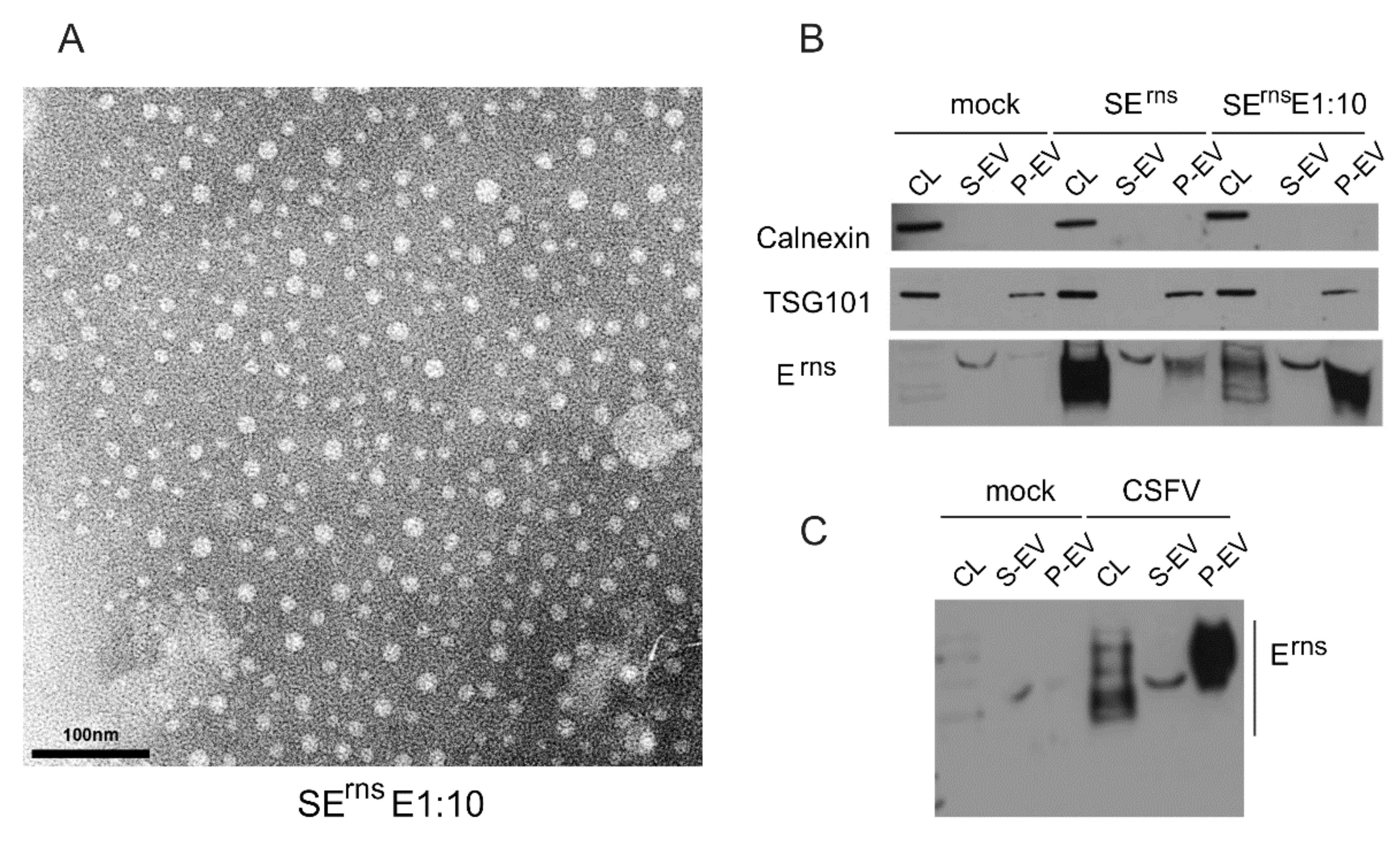
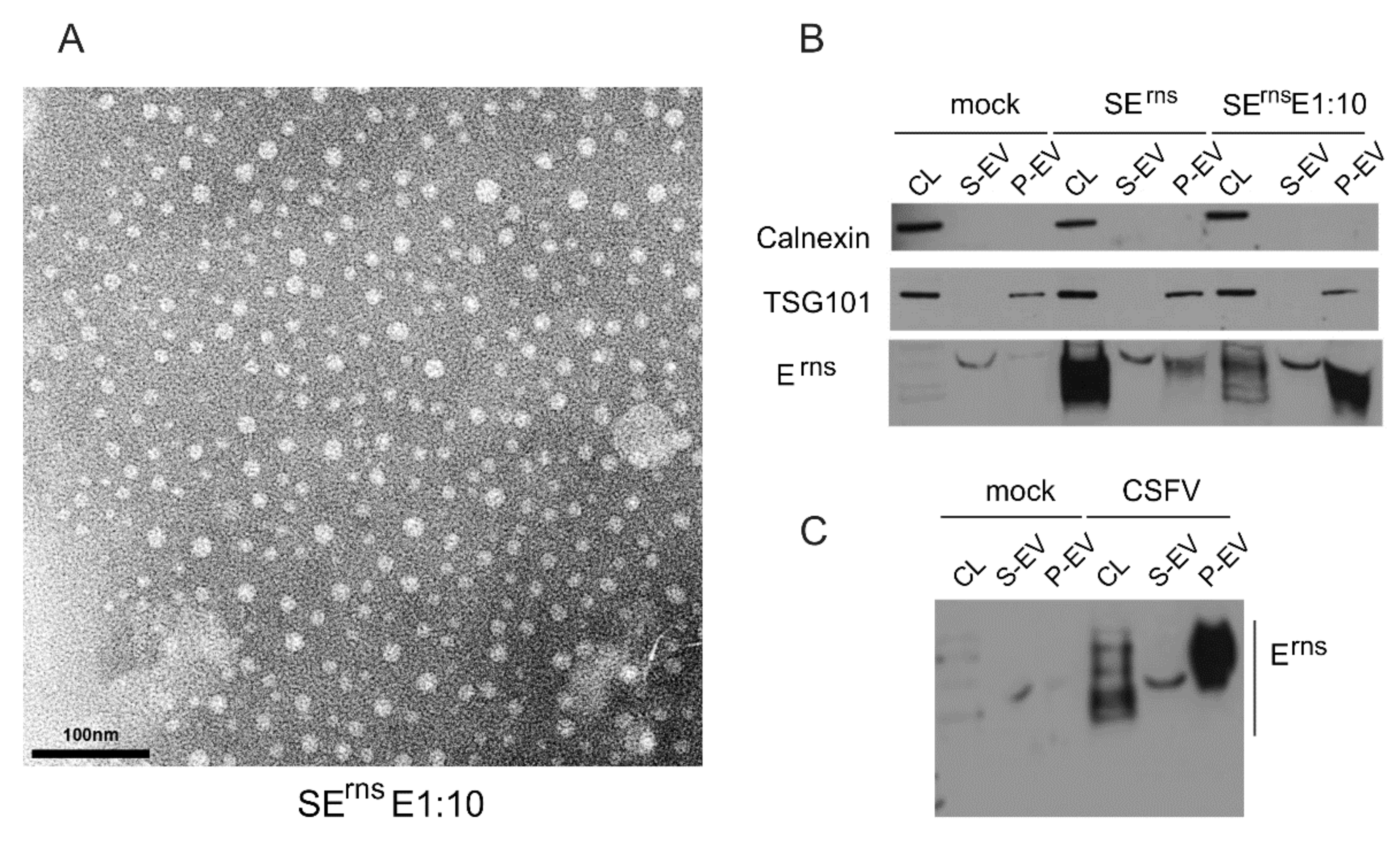
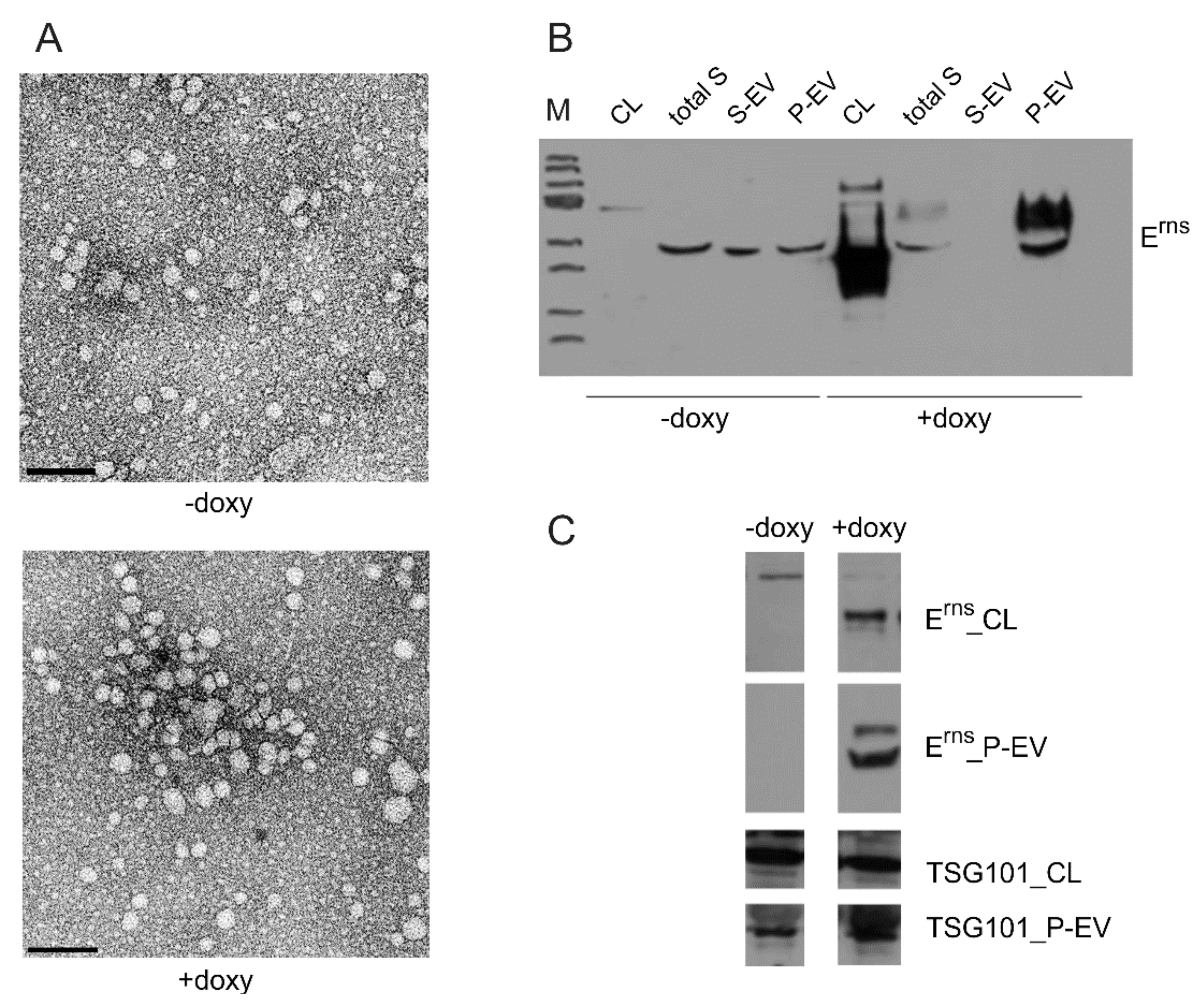
3.6.4. Erns Is Not Secreted as a Soluble Protein but as Part of Vesicles or Other Larger Structures
3.7. Effects of Erns Membrane Anchor Mutations on the Recovery and Viability of Infectious Viruses
3.8. Mutations of Certain Charged Amino Acids in the Erns Membrane Anchor Impair Virus Particle Formation or Release
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lindenbach, B.D.; Thiel, H.J.; Rice, C.M. Flaviviridae: The Viruses and Their Replication. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott—Raven Publishers: Philadelphia, PA, USA, 2007; Volume 5, pp. 1101–1152. [Google Scholar]
- Tautz, N.; Tews, B.A.; Meyers, G. The Molecular Biology of Pestiviruses. Adv. Virus Res. 2015, 93, 47–160. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Nie, Y.; Wang, P.; Ding, M.; Deng, H. Characterization of classical swine fever virus entry by using pseudotyped viruses: E1 and E2 are sufficient to mediate viral entry. Virology 2004, 330, 332–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronecker, S.; Zimmer, G.; Herrler, G.; Greiser-Wilke, I.; Grummer, B. Formation of bovine viral diarrhea virus E1–E2 heterodimers is essential for virus entry and depends on charged residues in the transmembrane domains. J. Gen. Virol. 2008, 89, 2114–2121. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Von Freyburg, M.; Elbers, K.; Meyers, G. Recovery of virulent and RNase-negative attenuated type 2 bovine viral diarrhea viruses from infectious cDNA clones. J. Virol. 2002, 76, 8494–8503. [Google Scholar] [CrossRef] [Green Version]
- Meyers, G.; Saalmüller, A.; Büttner, M. Mutations abrogating the RNase activity in glycoprotein e(rns) of the pestivirus classical swine fever virus lead to virus attenuation. J. Virol. 1999, 73, 10224–10235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tews, B.A.; Schürmann, E.M.; Meyers, G. Mutation of cysteine 171 of pestivirus E rns RNase prevents homodimer formation and leads to attenuation of classical swine fever virus. J. Virol. 2009, 83, 4823–4834. [Google Scholar] [CrossRef] [Green Version]
- Tucakov, A.K.; Yavuz, S.; Schurmann, E.M.; Mischler, M.; Klingebeil, A.; Meyers, G. Restoration of glycoprotein E(rns) dimerization via pseudoreversion partially restores virulence of classical swine fever virus. J. Gen. Virol. 2018, 99, 86–96. [Google Scholar] [CrossRef]
- Krey, T.; Bontems, F.; Vonrhein, C.; Vaney, M.C.; Bricogne, G.; Rümenapf, T.; Rey, F.A. Crystal structure of the pestivirus envelope glycoprotein E(rns) and mechanistic analysis of its ribonuclease activity. Structure 2012, 20, 862–873. [Google Scholar] [CrossRef] [Green Version]
- Schneider, R.; Unger, G.; Stark, R.; Schneider-Scherzer, E.; Thiel, H.J. Identification of a structural glycoprotein of an RNA virus as a ribonuclease. Science 1993, 261, 1169–1171. [Google Scholar] [CrossRef] [PubMed]
- Windisch, J.M.; Schneider, R.; Stark, R.; Weiland, E.; Meyers, G.; Thiel, H.J. RNase of Classical swine fever virus: Biochemical characterization and inhibition by virus-neutralizing monoclonal antibodies. J. Virol. 1996, 70, 352–358. [Google Scholar] [CrossRef] [Green Version]
- Hausmann, Y.; Roman-Sosa, G.; Thiel, H.J.; Rümenapf, T. Classical swine fever virus glycoprotein E rns is an endoribonuclease with an unusual base specificity. J. Virol. 2004, 78, 5507–5512. [Google Scholar] [CrossRef] [Green Version]
- Rümenapf, T.; Unger, G.; Strauss, J.H.; Thiel, H.J. Processing of the envelope glycoproteins of pestiviruses. J. Virol. 1993, 67, 3288–3295. [Google Scholar] [CrossRef] [Green Version]
- Tews, B.A.; Meyers, G. The Pestivirus Glycoprotein Erns Is Anchored in Plane in the Membrane via an Amphipathic Helix. J. Biol. Chem. 2007, 282, 32730–32741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luhtala, N.; Parker, R. T2 Family ribonucleases: Ancient enzymes with diverse roles. Trends Biochem. Sci. 2010, 35, 253–259. [Google Scholar] [CrossRef] [Green Version]
- Aberle, D.; Muhle-Goll, C.; Bürck, J.; Wolf, M.; Reisser, S.; Luy, B.; Wenzel, W.; Ulrich, A.S.; Meyers, G. Structure of the membrane anchor of pestivirus glycoprotein E(rns), a long tilted amphipathic helix. PLoS Pathog. 2014, 10, e1003973. [Google Scholar] [CrossRef] [PubMed]
- Aberle, D.; Oetter, K.M.; Meyers, G. Lipid Binding of the Amphipathic Helix Serving as Membrane Anchor of Pestivirus Glycoprotein Erns. PLoS ONE 2015, 10, e0135680. [Google Scholar] [CrossRef] [Green Version]
- Bintintan, I.; Meyers, G. A new type of signal peptidase cleavage site identified in an RNA virus polyprotein. J. Biol. Chem. 2010, 285, 8572–8584. [Google Scholar] [CrossRef] [Green Version]
- Mu, Y.; Bintintan, I.; Meyers, G. Downstream Sequences Control the Processing of the Pestivirus E(rns)-E1 Precursor. J. Virol. 2020, 95. [Google Scholar] [CrossRef] [PubMed]
- Magkouras, I.; Mätzener, P.; Rümenapf, T.; Peterhans, E.; Schweizer, M. RNase-dependent inhibition of extracellular, but not intracellular, dsRNA-induced interferon synthesis by Erns of pestiviruses. J. Gen. Virol. 2008, 89, 2501–2506. [Google Scholar] [CrossRef] [PubMed]
- Mätzener, P.; Magkouras, I.; Rümenapf, T.; Peterhans, E.; Schweizer, M. The viral RNase E(rns) prevents IFN type-I triggering by pestiviral single- and double-stranded RNAs. Virus Res. 2009, 140, 15–23. [Google Scholar] [CrossRef]
- Thiel, H.J.; Stark, R.; Weiland, E.; Rümenapf, T.; Meyers, G. Hog cholera virus: Molecular composition of virions from a pestivirus. J. Virol. 1991, 65, 4705–4712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyers, G.; Ege, A.; Fetzer, C.; von Freyburg, M.; Elbers, K.; Carr, V.; Prentice, H.; Charleston, B.; Schürmann, E.M. Bovine viral diarrhoea virus: Prevention of persistent foetal infection by a combination of two mutations affecting the Erns RNase and the Npro protease. J. Virol. 2007, 81, 3327–3338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reid, E.; Juleff, N.; Windsor, M.; Gubbins, S.; Roberts, L.; Morgan, S.; Meyers, G.; Perez-Martin, E.; Tchilian, E.; Charleston, B.; et al. Type I and III IFNs Produced by Plasmacytoid Dendritic Cells in Response to a Member of the Flaviviridae Suppress Cellular Immune Responses. J. Immunol. 2016, 196, 4214–4226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutter, G.; Ohlmann, M.; Erfle, V. Non-replicating vaccinia vector efficiently expresses bacteriophage T7 RNA polymerase. FEBS Lett. 1995, 371, 9–12. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, L.S.; Moss, B.; Rozenblatt, S. Replication-deficient vaccinia virus encoding bacteriophage T7 RNA polymerase for transient gene expression in mammalian cells. Virology 1995, 210, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Krey, T.; Thiel, H.J.; Rümenapf, T. Acid-resistant bovine pestivirus requires activation for pH-triggered fusion during entry. J. Virol. 2005, 79, 4191–4200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krey, T.; Himmelreich, A.; Heimann, M.; Menge, C.; Thiel, H.J.; Maurer, K.; Rümenapf, T. Function of bovine CD46 as a cellular receptor for bovine viral diarrhea virus is determined by complement control protein 1. J. Virol. 2006, 80, 3912–3922. [Google Scholar] [CrossRef] [Green Version]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Weiland, E.; Ahl, R.; Stark, R.; Weiland, F.; Thiel, H.J. A second envelope glycoprotein mediates neutralization of a pestivirus, hog cholera virus. J. Virol. 1992, 66, 3677–3682. [Google Scholar] [CrossRef] [Green Version]
- Schägger, H.; Jagow, G.v. Tricine- sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 1987, 166, 368–379. [Google Scholar] [CrossRef]
- Meyers, G.; Thiel, H.J.; Rümenapf, T. Classical swine fever virus: Recovery of infectious viruses from cDNA constructs and generation of recombinant cytopathogenic defective interfering particles. J. Virol. 1996, 70, 1588–1595. [Google Scholar] [CrossRef] [Green Version]
- Weiland, E.; Stark, R.; Haas, B.; Rümenapf, T.; Meyers, G.; Thiel, H.J. Pestivirus glycoprotein which induces neutralizing antibodies forms part of a disulfide linked heterodimer. J. Virol. 1990, 64, 3563–3569. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, B.; Beer, M.; Schelp, C.; Schirrmeier, H.; Depner, K. Validation of a real-time RT-PCR assay for sensitive and specific detection of classical swine fever. J. Virol. Methods 2005, 130, 36–44. [Google Scholar] [CrossRef]
- Hoffmann, B.; Depner, K.; Schirrmeier, H.; Beer, M. A universal heterologous internal control system for duplex real-time RT-PCR assays used in a detection system for pestiviruses. J. Virol. Methods 2006, 136, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Fetzer, C.; Tews, B.A.; Meyers, G. The carboxy-terminal sequence of the pestivirus glycoprotein E(rns) represents an unusual type of membrane anchor. J. Virol. 2005, 79, 11901–11913. [Google Scholar] [CrossRef] [Green Version]
- Burrack, S.; Aberle, D.; Bürck, J.; Ulrich, A.S.; Meyers, G. A new type of intracellular retention signal identified in a pestivirus structural glycoprotein. FASEB J. 2012, 26, 3292–3305. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Murray, C.L.; Thiel, H.J.; Rice, C.M. Flaviviridae. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott—Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 6, pp. 712–746. [Google Scholar]
- Dubuisson, J. Hepatitis C virus proteins. World J. Gastroenterol. 2007, 13, 2406–2415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartenschlager, R.; Ahlborn-Laake, L.; Mous, J.; Jacobsen, H. Kinetic and structural analyses of hepatitis C virus polyprotein processing. J. Virol. 1994, 68, 5045–5055. [Google Scholar] [CrossRef] [Green Version]
- Carrere-Kremer, S.; Montpellier, C.; Lorenzo, L.; Brulin, B.; Cocquerel, L.; Belouzard, S.; Penin, F.; Dubuisson, J. Regulation of hepatitis C virus polyprotein processing by signal peptidase involves structural determinants at the p7 sequence junctions. J. Biol. Chem. 2004, 279, 41384–41392. [Google Scholar] [CrossRef] [Green Version]
- Cocquerel, L.; Op De, B.A.; Lambot, M.; Roussel, J.; Delgrange, D.; Pillez, A.; Wychowski, C.; Penin, F.; Dubuisson, J. Topological changes in the transmembrane domains of hepatitis C virus envelope glycoproteins. EMBO J. 2002, 21, 2893–2902. [Google Scholar] [CrossRef] [Green Version]
- Heimann, M.; Roman-Sosa, G.; Martoglio, B.; Thiel, H.J.; Rümenapf, T. Core protein of pestiviruses is processed at the C terminus by signal peptide peptidase. J. Virol. 2006, 80, 1915–1921. [Google Scholar] [CrossRef] [Green Version]
- Hussy, P.; Langen, H.; Mous, J.; Jacobsen, H. Hepatitis C virus core protein: Carboxy-terminal boundaries of two processed species suggest cleavage by a signal peptide peptidase. Virology 1996, 224, 93–104. [Google Scholar] [CrossRef] [Green Version]
- McLauchlan, J.; Lemberg, M.K.; Hope, G.; Martoglio, B. Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. EMBO J. 2002, 21, 3980–3988. [Google Scholar] [CrossRef]
- Wegelt, A.; Reimann, I.; Zemke, J.; Beer, M. New insights into processing of bovine viral diarrhea virus glycoproteins E(rns) and E1. J. Gen. Virol. 2009, 90, 2462–2467. [Google Scholar] [CrossRef]
- Baker, R.K.; Bentivoglio, G.P.; Lively, M.O. Partial purification of microsomal signal peptidase from hen oviduct. J. Cell Biochem. 1986, 32, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.K.; Lively, M.O. Purification and characterization of hen oviduct microsomal signal peptidase. Biochemistry 1987, 26, 8561–8567. [Google Scholar] [CrossRef] [PubMed]
- Lively, M.O. Signal peptidases in protein biosynthesis and intracellular transport. Curr. Opin. Cell Biol. 1989, 1, 1188–1193. [Google Scholar] [CrossRef]
- Lively, M.O.; Newsome, A.L.; Nusier, M. Eukaryote microsomal signal peptidases. Methods Enzymol. 1994, 244, 301–314. [Google Scholar]
- Snapp, E.L.; McCaul, N.; Quandte, M.; Cabartova, Z.; Bontjer, I.; Kallgren, C.; Nilsson, I.; Land, A.; von Heijne, G.; Sanders, R.W.; et al. Structure and topology around the cleavage site regulate post-translational cleavage of the HIV-1 gp160 signal peptide. eLife 2017, 6. [Google Scholar] [CrossRef]
- Von Heijne, G. Signal sequences are not uniformly hydrophobic. J. Mol. Biol. 1982, 159, 537–541. [Google Scholar] [CrossRef]
- Von Heijne, G. The signal peptide. J. Membr. Biol. 1990, 115, 195–201. [Google Scholar] [CrossRef]
- Rehm, A.; Stern, P.; Ploegh, H.L.; Tortorella, D. Signal peptide cleavage of a type I membrane protein, HCMV US11, is dependent on its membrane anchor. EMBO J. 2001, 20, 1573–1582. [Google Scholar] [CrossRef] [Green Version]
- Von Heijne, G. Signal sequences. The limits of variation. J. Mol. Biol. 1985, 184, 99–105. [Google Scholar] [CrossRef]
- Hegde, R.S.; Bernstein, H.D. The surprising complexity of signal sequences. Trends Biochem. Sci. 2006, 31, 563–571. [Google Scholar] [CrossRef]
- Mu, Y.; Radtke, C.; Tews, B.A.; Meyers, G. Characterization of membrane topology and retention signal of pestiviral glycoprotein E1. J. Virol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Lippincott-Schwartz, J.; Yuan, L.C.; Bonifacino, J.S.; Klausner, R.D. Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: Evidence for membrane cycling from Golgi to ER. Cell 1989, 56, 801–813. [Google Scholar] [CrossRef]
- Oda, K.; Hirose, S.; Takami, N.; Misumi, Y.; Takatsuki, A.; Ikehara, Y. Brefeldin A arrests the intracellular transport of a precursor of complement C3 before its conversion site in rat hepatocytes. FEBS Lett. 1987, 214, 135–138. [Google Scholar] [CrossRef] [Green Version]
- Jackson, R.C.; Blobel, G. Post-translational cleavage of presecretory proteins with an extract of rough microsomes from dog pancreas containing signal peptidase activity. Proc. Natl. Acad. Sci. USA 1977, 74, 5598–5602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, R.C.; Blobel, G. Post-translational processing of full-length presecretory proteins with canine pancreatic signal peptidase. Ann. N. Y. Acad. Sci. 1980, 343, 391–404. [Google Scholar] [CrossRef]
- Casanova, C.L.; Xue, G.; Taracha, E.L.; Dobbelaere, D.A. Post-translational signal peptide cleavage controls differential epitope recognition in the QP-rich domain of recombinant Theileria parva PIM. Mol. Biochem. Parasitol. 2006, 149, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, I.; Johnson, A.E.; von Heijne, G. Cleavage of a tail-anchored protein by signal peptidase. FEBS Lett. 2002, 516, 106–108. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, I.; Johnson, A.E.; von Heijne, G. How hydrophobic is alanine? J. Biol. Chem. 2003, 278, 29389–29393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Geest, M.; Nilsson, I.; von Heijne, G.; Lolkema, J.S. Insertion of a bacterial secondary transport protein in the endoplasmic reticulum membrane. J. Biol. Chem. 1999, 274, 2816–2823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karla, A.; Lively, M.O.; Paetzel, M.; Dalbey, R. The identification of residues that control signal peptidase cleavage fidelity and substrate specificity. J. Biol. Chem. 2005, 280, 6731–6741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martoglio, B.; Dobberstein, B. Signal sequences: More than just greasy peptides. Trends Cell Biol. 1998, 8, 410–415. [Google Scholar] [CrossRef]
- Robson, A.; Collinson, I. The structure of the Sec complex and the problem of protein translocation. EMBO Rep. 2006, 7, 1099–1103. [Google Scholar] [CrossRef] [PubMed]
- Von Heijne, G. Cleavage-site motifs in protein targeting sequences. Genet. Eng. 1992, 14, 1–11. [Google Scholar]
- Von Heijne, G. The membrane protein universe: What’s out there and why bother? J. Intern. Med. 2007, 261, 543–557. [Google Scholar] [CrossRef] [PubMed]
- White, S.H.; von Heijne, G. Do protein-lipid interactions determine the recognition of transmembrane helices at the ER translocon? Biochem. Soc. Trans. 2005, 33, 1012–1015. [Google Scholar] [CrossRef] [Green Version]
- White, S.H.; von Heijne, G. Transmembrane helices before, during, and after insertion. Curr. Opin. Struct. Biol. 2005, 15, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Yamshchikov, V.F.; Compans, R.W. Processing of the intracellular form of the west Nile virus capsid protein by the viral NS2B-NS3 protease: An in vitro study. J. Virol. 1994, 68, 5765–5771. [Google Scholar] [CrossRef] [Green Version]
- Drin, G.; Antonny, B. Cell biology: Helices sculpt membrane. Nature 2005, 437, 1247–1249. [Google Scholar] [CrossRef]
- Drin, G.; Antonny, B. Amphipathic helices and membrane curvature. FEBS Lett. 2010, 584, 1840–1847. [Google Scholar] [CrossRef] [PubMed]
- Prevost, C.; Sharp, M.E.; Kory, N.; Lin, Q.; Voth, G.A.; Farese, R.V., Jr.; Walther, T.C. Mechanism and Determinants of Amphipathic Helix-Containing Protein Targeting to Lipid Droplets. Dev. Cell 2018, 44, 73–86.e74. [Google Scholar] [CrossRef] [PubMed]
- Zhukovsky, M.A.; Filograna, A.; Luini, A.; Corda, D.; Valente, C. Protein Amphipathic Helix Insertion: A Mechanism to Induce Membrane Fission. Front. Cell Dev. Biol. 2019, 7, 291. [Google Scholar] [CrossRef]
- Walther, T.H.; Gottselig, C.; Grage, S.L.; Wolf, M.; Vargiu, A.V.; Klein, M.J.; Vollmer, S.; Prock, S.; Hartmann, M.; Afonin, S.; et al. Folding and self-assembly of the TatA translocation pore based on a charge zipper mechanism. Cell 2013, 152, 316–326. [Google Scholar] [CrossRef] [Green Version]
- Walther, T.H.; Ulrich, A.S. Transmembrane helix assembly and the role of salt bridges. Curr. Opin. Struct. Biol. 2014, 27, 63–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oetter, K.M.; Kühn, J.; Meyers, G. Charged Residues in the Membrane Anchor of the Pestiviral E(rns) Protein Are Important for Processing and Secretion of E(rns) and Recovery of Infectious Viruses. Viruses 2021, 13, 444. [Google Scholar] [CrossRef] [PubMed]
- Amberg, S.M.; Nestorowicz, A.; McCourt, D.W.; Rice, C.M. NS2B-3 proteinase-mediated processing in the yellow fever virus structural region: In vitro and in vivo studies. J. Virol. 1994, 68, 3794–3802. [Google Scholar] [CrossRef] [Green Version]
- Lobigs, M. Flavivirus premembrane protein cleavage and spike heterodimer secretion require the function of the viral proteinase NS3. Proc. Natl. Acad. Sci. USA 1993, 90, 6218–6222. [Google Scholar] [CrossRef] [Green Version]
- Stocks, C.E.; Lobigs, M. Signal peptidase cleavage at the flavivirus C-prM junction: Dependence on the viral NS2B-3 protease for efficient processing requires determinants in C, the signal peptide, and prM. J. Virol. 1998, 72, 2141–2149. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Stocks, C.E.; Amberg, S.M.; Rice, C.M.; Lobigs, M. Mutagenesis of the signal sequence of yellow fever virus prM protein: Enhancement of signalase cleavage In vitro is lethal for virus production. J. Virol. 2000, 74, 24–32. [Google Scholar] [CrossRef] [Green Version]
- Earp, L.J.; Delos, S.E.; Park, H.E.; White, J.M. The many mechanisms of viral membrane fusion proteins. Curr. Top. Microbiol. Immunol. 2005, 285, 25–66. [Google Scholar] [CrossRef]
- Harrison, S.C. Viral membrane fusion. Virology 2015, 479–480, 498–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kielian, M. Mechanisms of Virus Membrane Fusion Proteins. Annu. Rev. Virol. 2014, 1, 171–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rey, F.A.; Lok, S.M. Common Features of Enveloped Viruses and Implications for Immunogen Design for Next-Generation Vaccines. Cell 2018, 172, 1319–1334. [Google Scholar] [CrossRef] [PubMed]
- El Omari, K.; Iourin, O.; Harlos, K.; Grimes, J.M.; Stuart, D.I. Structure of a pestivirus envelope glycoprotein E2 clarifies its role in cell entry. Cell Rep. 2013, 3, 30–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holinka, L.G.; Largo, E.; Gladue, D.P.; O’Donnell, V.; Risatti, G.R.; Nieva, J.L.; Borca, M.V. Alteration of a Second Putative Fusion Peptide of Structural Glycoprotein E2 of Classical Swine Fever Virus Alters Virus Replication and Virulence in Swine. J. Virol. 2016, 90, 10299–10308. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Modis, Y. A novel membrane fusion protein family in Flaviviridae? Trends Microbiol. 2014, 22, 176–182. [Google Scholar] [CrossRef]
- Li, Y.; Wang, J.; Kanai, R.; Modis, Y. Crystal structure of glycoprotein E2 from bovine viral diarrhea virus. Proc. Natl. Acad. Sci. USA 2013, 110, 6805–6810. [Google Scholar] [CrossRef] [Green Version]
- Lavie, M.; Goffard, A.; Dubuisson, J. HCV Glycoproteins: Assembly of a Functional E1–E2 Heterodimer. In Hepatitis C Viruses: Genomes and Molecular Biology; Tan, S.L., Ed.; Horizon Bioscience: Norfolk, UK, 2006. [Google Scholar]
- Vieyres, G.; Dubuisson, J.; Pietschmann, T. Incorporation of hepatitis C virus E1 and E2 glycoproteins: The keystones on a peculiar virion. Viruses 2014, 6, 1149–1187. [Google Scholar] [CrossRef] [Green Version]
- Reimann, I.; Meyers, G.; Beer, M. Trans-complementation of autonomously replicating Bovine viral diarrhea virus replicons with deletions in the E2 coding region. Virology 2003, 307, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Reimann, I.; Semmler, I.; Beer, M. Packaged replicons of bovine viral diarrhea virus are capable of inducing a protective immune response. Virology 2007, 366, 377–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmeiser, S.; Mast, J.; Thiel, H.J.; König, M. Morphogenesis of pestiviruses: New insights from ultrastructural studies of strain Giraffe-1. J. Virol. 2014, 88, 2717–2724. [Google Scholar] [CrossRef] [Green Version]
- Weiland, F.; Weiland, E.; Unger, G.; Saalmüller, A.; Thiel, H.J. Localization of pestiviral envelope proteins E(rns) and E2 at the cell surface and on isolated particles. J. Gen. Virol. 1999, 80, 1157–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grummer, B.; Beer, M.; Liebler-Tenorio, E.; Greiser-Wilke, I. Localization of viral proteins in cells infected with bovine viral diarrhoea virus. J. Gen. Virol. 2001, 82, 2597–2605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciczora, Y.; Callens, N.; Montpellier, C.; Bartosch, B.; Cosset, F.L.; Op De, B.A.; Dubuisson, J. Contribution of the charged residues of hepatitis C virus glycoprotein E2 transmembrane domain to the functions of the E1E2 heterodimer. J. Gen. Virol. 2005, 86, 2793–2798. [Google Scholar] [CrossRef]
- Cocquerel, L.; Duvet, S.; Meunier, J.C.; Pillez, A.; Cacan, R.; Wychowski, C.; Dubuisson, J. The transmembrane domain of hepatitis C virus glycoprotein E1 is a signal for static retention in the endoplasmic reticulum. J. Virol. 1999, 73, 2641–2649. [Google Scholar] [CrossRef] [Green Version]
- Cocquerel, L.; Meunier, J.C.; Pillez, A.; Wychowski, C.; Dubuisson, J. A retention signal necessary and sufficient for endoplasmic reticulum localization maps to the transmembrane domain of hepatitis C virus glycoprotein E2. J. Virol. 1998, 72, 2183–2191. [Google Scholar] [CrossRef] [Green Version]
- Radtke, C.; Tews, B.A. Retention and topology of the bovine viral diarrhea virus glycoprotein E2. J. Gen. Virol. 2017, 98, 2482–2494. [Google Scholar] [CrossRef]
- Köhl, W.; Zimmer, G.; Greiser-Wilke, I.; Haas, L.; Moennig, V.; Herrler, G. The surface glycoprotein E2 of bovine viral diarrhoea virus contains an intracellular localization signal. J. Gen. Virol. 2004, 85, 1101–1111. [Google Scholar] [CrossRef]
- Ciczora, Y.; Callens, N.; Seron, K.; Rouille, Y.; Dubuisson, J. Identification of a dominant endoplasmic reticulum-retention signal in yellow fever virus pre-membrane protein. J. Gen. Virol. 2010, 91, 404–414. [Google Scholar] [CrossRef]
- Vander Heyden, A.B.; Naismith, T.V.; Snapp, E.L.; Hanson, P.I. Static retention of the lumenal monotopic membrane protein torsinA in the endoplasmic reticulum. EMBO J. 2011, 30, 3217–3231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciglic, M.I.; Jackson, P.J.; Raillard, S.A.; Haugg, M.; Jermann, T.M.; Opitz, J.G.; Trabesinger-Ruf, N.; Benner, S.A. Origin of dimeric structure in the ribonuclease superfamily. Biochemistry 1998, 37, 4008–4022. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Raines, R.T. A misfolded but active dimer of bovine seminal ribonucleaseion. Eur. J. Biochem. 1994, 224, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Soucek, J.; Matousek, J.; Raines, R.T. Mechanism of ribonuclease cytotoxicity. J. Biol. Chem. 1995, 270, 31097–31102. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S.; Soucek, J.; Matousek, J.; Raines, R.T. Catalytic activity of bovine seminal ribonuclease is essential for its immunosuppressive and other biological activities. Biochem.J. 1995, 308, 547–550. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Raines, R.T. Cytotoxicity of bovine seminal ribonuclease: Monomer versus dimer. Biochemistry 2005, 44, 15760–15767. [Google Scholar] [CrossRef]
- Lazar, C.; Zitzmann, N.; Dwek, R.A.; Branza-Nichita, N. The pestivirus E(rns) glycoprotein interacts with E2 in both infected cells and mature virions. Virology 2003, 314, 696–705. [Google Scholar] [CrossRef] [Green Version]
- Maurer, K.; Krey, T.; Moennig, V.; Thiel, H.J.; Rümenapf, T. CD46 is a cellular receptor for bovine viral diarrhea virus. J. Virol. 2004, 78, 1792–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, T.; Tautz, N.; Thiel, H.J. E2-p7 region of the bovine viral diarrhea virus polyprotein: Processing and functional studies. J. Virol. 2000, 74, 9498–9506. [Google Scholar] [CrossRef] [Green Version]
- Teasdale, R.D.; Jackson, M.R. Signal-mediated sorting of membrane proteins between the endoplasmic reticulum and the golgi apparatus. Annu. Rev. Cell Dev. Biol. 1996, 12, 27–54. [Google Scholar] [CrossRef]
- Yamamoto, K.; Fujii, R.; Toyofuku, Y.; Saito, T.; Koseki, H.; Hsu, V.W.; Aoe, T. The KDEL receptor mediates a retrieval mechanism that contributes to quality control at the endoplasmic reticulum. EMBO J. 2001, 20, 3082–3091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Thery, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell. Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Segrest, J.P.; De Loof, H.; Dohlman, J.G.; Brouillette, C.G.; Anantharamaiah, G.M. Amphipathic helix motif: Classes and properties. Proteins 1990, 8, 103–117. [Google Scholar] [CrossRef]
- Hulst, M.M.; Moormann, R.J. Erns protein of pestiviruses. Methods Enzymol. 2001, 342, 431–440. [Google Scholar]
- Hulst, M.M.; van Gennip, H.G.; Vlot, A.C.; Schooten, E.; de Smit, A.J.; Moormann, R.J. Interaction of classical swine fever virus with membrane-associated heparan sulfate: Role for virus replication in vivo and virulence. J. Virol. 2001, 75, 9585–9595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, M.; Flick-Smith, H.; McCauley, J.W. Interactions of bovine viral diarrhoea virus glycoprotein E(rns) with cell surface glycosaminoglycans. J. Gen. Virol. 2000, 81, 451–459. [Google Scholar]
- Iqbal, M.; McCauley, J.W. Identification of the glycosaminoglycan-binding site on the glycoprotein E(rns) of bovine viral diarrhoea virus by site-directed mutagenesis. J. Gen. Virol. 2002, 83, 2153–2159. [Google Scholar] [CrossRef]
- Sainz, I.F.; Holinka, L.G.; Lu, Z.; Risatti, G.R.; Borca, M.V. Removal of a N-linked glycosylation site of classical swine fever virus strain Brescia Erns glycoprotein affects virulence in swine. Virology 2008, 370, 122–129. [Google Scholar] [CrossRef] [Green Version]
- Zürcher, C.; Sauter, K.S.; Mathys, V.; Wyss, F.; Schweizer, M. Prolonged activity of the pestiviral RNase Erns as an interferon antagonist after uptake by clathrin-mediated endocytosis. J. Virol. 2014, 88, 7235–7243. [Google Scholar] [CrossRef] [Green Version]
- Zürcher, C.; Sauter, K.S.; Schweizer, M. Pestiviral E(rns) blocks TLR-3-dependent IFN synthesis by LL37 complexed RNA. Vet. Microbiol. 2014, 174, 399–408. [Google Scholar] [CrossRef]
- Lussi, C.; Sauter, K.S.; Schweizer, M. Homodimerisation-independent cleavage of dsRNA by a pestiviral nicking endoribonuclease. Sci. Rep. 2018, 8, 8226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulst, M.M.; Moormann, R.J. Inhibition of pestivirus infection in cell culture by envelope proteins E(rns) and E2 of classical swine fever virus: E(rns) and E2 interact with different receptors. J. Gen. Virol. 1997, 78, 2779–2787. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Krabben, L.; Wang, F.; Veit, M. Glycoprotein 3 of Porcine Reproductive and Respiratory Syndrome Virus Exhibits an Unusual Hairpin-Like Membrane Topology. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, R.J.; Jensen, S.S.; Lim, J.W. Proteomic profiling of exosomes: Current perspectives. Proteomics 2008, 8, 4083–4099. [Google Scholar] [CrossRef]
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Hawari, F.I.; Rouhani, F.N.; Cui, X.; Yu, Z.X.; Buckley, C.; Kaler, M.; Levine, S.J. Release of full-length 55-kDa TNF receptor 1 in exosome-like vesicles: A mechanism for generation of soluble cytokine receptors. Proc. Natl. Acad. Sci. USA 2004, 101, 1297–1302. [Google Scholar] [CrossRef] [Green Version]
- Thery, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Bauhofer, O.; Summerfield, A.; Sakoda, Y.; Tratschin, J.D.; Hofmann, M.A.; Ruggli, N. Classical swine fever virus Npro interacts with interferon regulatory factor 3 and induces its proteasomal degradation. J. Virol. 2007, 81, 3087–3096. [Google Scholar] [CrossRef] [Green Version]
- Doceul, V.; Charleston, B.; Crooke, H.; Reid, E.; Powell, P.P.; Seago, J. The Npro product of classical swine fever virus interacts with IκBα, the NF-κB inhibitor. J. Gen. Virol. 2008, 89, 1881–1889. [Google Scholar] [CrossRef]
- Fiebach, A.R.; Guzylack-Piriou, L.; Python, S.; Summerfield, A.; Ruggli, N. Classical Swine Fever virus npro limits type I interferon induction in plasmacytoid dendritic cells by interacting with interferon regulatory factor 7. J. Virol. 2011, 85, 8002–8011. [Google Scholar] [CrossRef] [Green Version]
- Hilton, L.; Moganeradj, K.; Zhang, G.; Chen, Y.H.; Randall, R.E.; McCauley, J.W.; Goodbourn, S. The NPro product of bovine viral diarrhea virus inhibits DNA binding by interferon regulatory factor 3 and targets it for proteasomal degradation. J. Virol. 2006, 80, 11723–11732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Rocca, S.A.; Herbert, R.J.; Crooke, H.; Drew, T.W.; Wileman, T.E.; Powell, P.P. Loss of interferon regulatory factor 3 in cells infected with classical swine fever virus involves the N-terminal protease, Npro. J. Virol. 2005, 79, 7239–7247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seago, J.; Hilton, L.; Reid, E.; Doceul, V.; Jeyatheesan, J.; Moganeradj, K.; McCauley, J.; Charleston, B.; Goodbourn, S. The Npro product of classical swine fever virus and bovine viral diarrhea virus uses a conserved mechanism to target interferon regulatory factor-3. J. Gen. Virol. 2007, 88, 3002–3006. [Google Scholar] [CrossRef]
- Tamura, T.; Nagashima, N.; Ruggli, N.; Summerfield, A.; Kida, H.; Sakoda, Y. Npro of classical swine fever virus contributes to pathogenicity in pigs by preventing type I interferon induction at local replication sites. Vet. Res. 2014, 45, 47. [Google Scholar] [CrossRef] [Green Version]
- Python, S.; Gerber, M.; Suter, R.; Ruggli, N.; Summerfield, A. Efficient sensing of infected cells in absence of virus particles by plasmacytoid dendritic cells is blocked by the viral ribonuclease E(rns.). PLoS Pathog. 2013, 9, e1003412. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
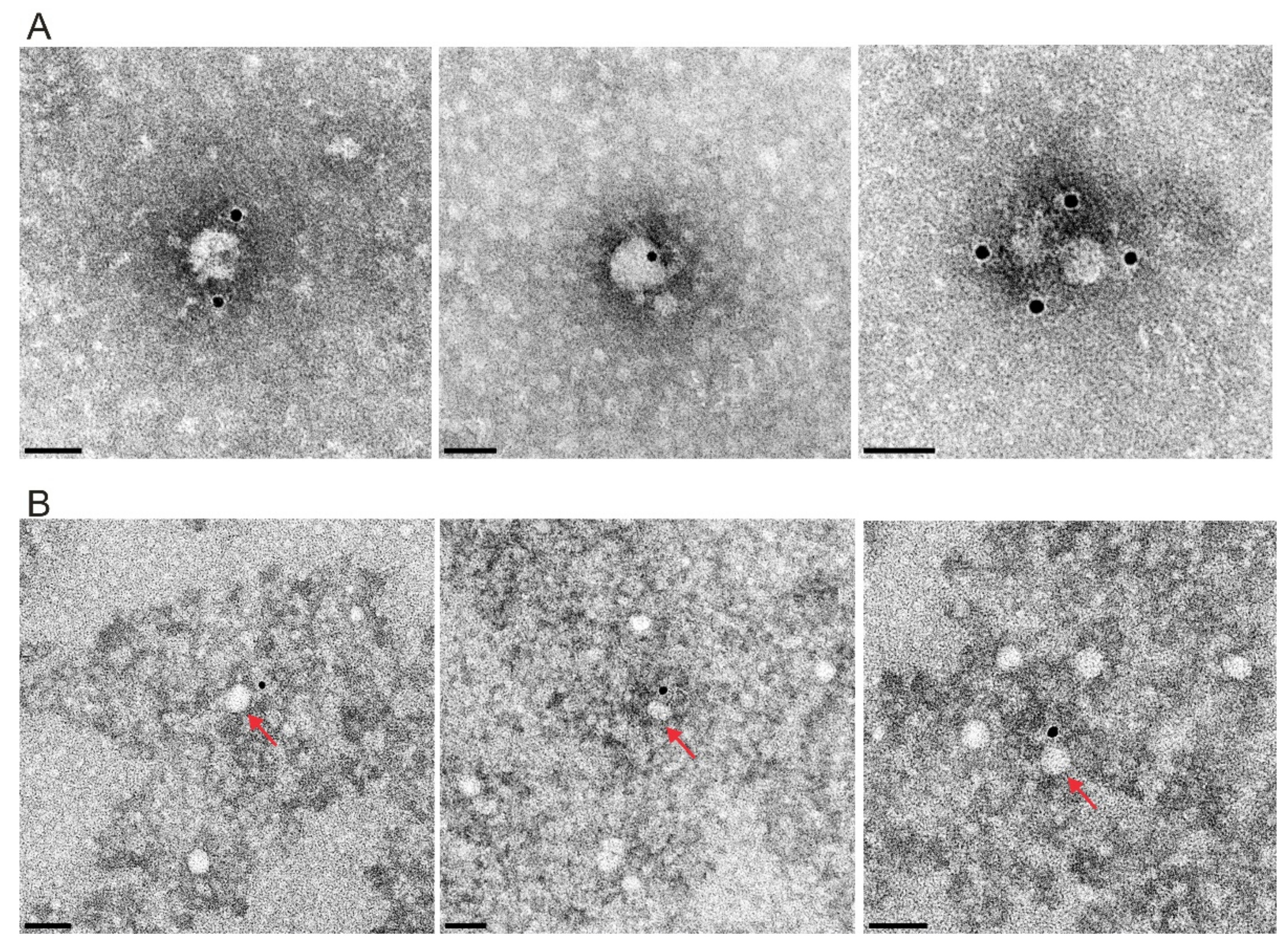{kind=link}
{kind=link}
{kind=link}
| Construct | Mutation | Charge | Precursor | Secretion | Replicon | Inf. Virus | Seq | Charge Rec. Virus |
|---|---|---|---|---|---|---|---|---|
| wt | - | ---/+++ | 14% | 7% | yes | yes | wt | ---/+++ |
| ΔErns | deletion of Erns gene | n.a. | n.a. | n.a. | yes | no | n.a. | n.a. |
| 12 | D7R/D4R/R9E | ++-/+-+ | 19% | 29% | yes | no | n.a. | n.a. |
| 18 | D7R/D4R/E1R/R4E/R9E/R6E | +++/--- | 30% | 43% | yes | no | n.a. | n.a. |
| 27 | R4E/R9E | ---/--+ | 44% | 63% | yes | yes | 4K/9R | ---/+++ |
| 29 | D7R | +--/+++ | 24% | 5% | yes | yes | 7R | +--/+++ |
| 30 | D4R | -+-/+++ | 23% | 26% | yes | no | n.a. | n.a. |
| 31 | E1R | --+/+++ | 16% | 28% | yes | no | n.a. | n.a. |
| 32 | R4E | ---/-++ | 41% | 26% | yes | yes | 4K or 4R | ---/+++ |
| 33 | R9E | ---/+-+ | 21% | 29% | yes | yes | 9E | ---/+-+ |
| 34 | R6E | ---/++- | 22% | 28% | yes | yes | 6K | ---/++- |
| 50 | D7a | a--/+++ | 14% | n.t. | yes | yes | 7a | a--/+++ |
| 51 | D4a | -a-/+++ | 16% | n.t. | yes | yes | 4D | ---/+++ |
| 35 | E1a | --a/+++ | 12% | n.t. | yes | yes | 1E | ---/+++ |
| 36 | R4a | ---/a++ | 19% | n.t. | yes | yes | 4R | ---/+++ |
| 52 | R9a | ---/+a+ | 16% | n.t. | yes | yes | 9a | ---/+a+ |
| 53 | R6a | ---/++a | n.t. | n.t. | yes | yes | 6a | ---/++a |
| E1/3 | P497A | n.a. | n.t. | n.t. | yes | yes | 497A | n.a. |
| E1/4 | Y498T | n.a. | n.t. | n.t. | yes | yes | 498Y | n.a. |
| E1/5 | C499S | n.a. | n.t. | n.t. | yes | yes | 499C | n.a. |
| E1/3.4.5 | PYC497-499AAA | n.a. | n.t. | n.t. | yes | no | n.a. | n.a. |
| E1/1.2.3.+ | LSP495-497EQKL | n.a. | n.t. | n.t. | yes | yes | EQKP | n.a. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tews, B.A.; Klingebeil, A.; Kühn, J.; Franzke, K.; Rümenapf, T.; Meyers, G. The Erns Carboxyterminus: Much More Than a Membrane Anchor. Viruses 2021, 13, 1203. https://doi.org/10.3390/v13071203
Tews BA, Klingebeil A, Kühn J, Franzke K, Rümenapf T, Meyers G. The Erns Carboxyterminus: Much More Than a Membrane Anchor. Viruses. 2021; 13(7):1203. https://doi.org/10.3390/v13071203
Chicago/Turabian StyleTews, Birke Andrea, Anne Klingebeil, Juliane Kühn, Kati Franzke, Till Rümenapf, and Gregor Meyers. 2021. "The Erns Carboxyterminus: Much More Than a Membrane Anchor" Viruses 13, no. 7: 1203. https://doi.org/10.3390/v13071203
APA StyleTews, B. A., Klingebeil, A., Kühn, J., Franzke, K., Rümenapf, T., & Meyers, G. (2021). The Erns Carboxyterminus: Much More Than a Membrane Anchor. Viruses, 13(7), 1203. https://doi.org/10.3390/v13071203