
How Do Enveloped Viruses Exploit the Secretory Proprotein Convertases to Regulate Infectivity and Spread?


Abstract
:1. Introduction
2. Proprotein Convertases and Enveloped Viruses
2.1. Furin in Viral Infections and Pathogenicity
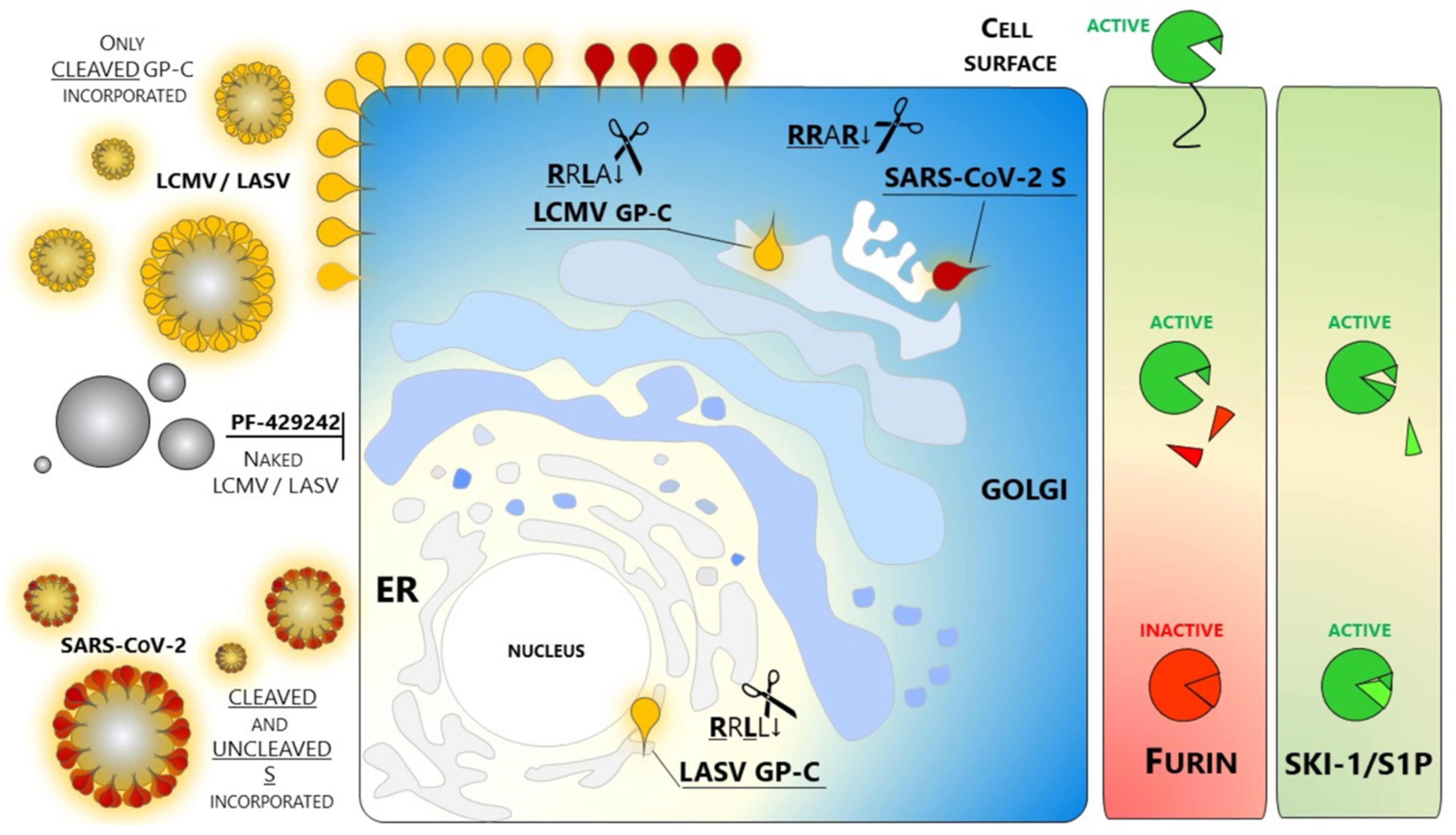
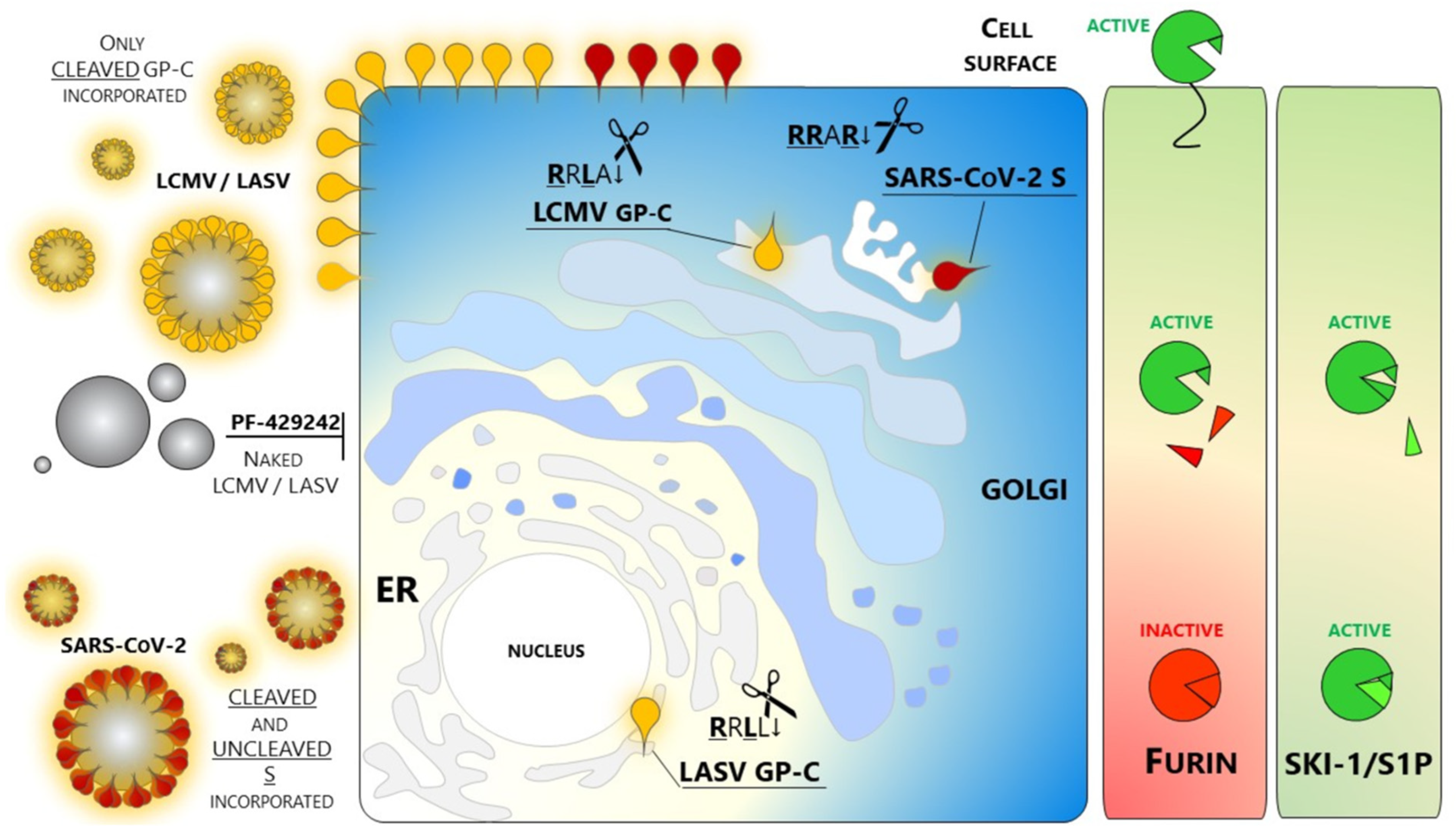
2.2. Coronavirus Infections, Including SARS-CoV-2
2.3. PCSK9 and Viral Infections
2.4. Implications of SKI-1/S1P in Viral Infections
- Non-peptide small molecules: Because of their properties and stability, this class of inhibitors are generally preferred over others for in vivo use. A small molecule SKI-1/S1P inhibitor PF-429242 was developed by Pfizer [136,137] and tested as an antiviral targeting GP-C processing and productive infection of arenaviruses. SKI-1/S1P inhibition by PF-429242 suppresses viral replication in cells infected with LASV, LCMV [138], and New World arenaviruses [139]. Interruption of drug treatment did not result in re-emergence of infection, indicating that PF-429242 treatment leads to virus extinction. Of note, Stefan Kunz found that the drug is capable of clearing LCMV from chronically infected cells with no emergence of escape variants [139]. This finding is intriguing since an LCMV mutant engineered to carry the RRRR↓ mutation is viable and fit [135]. The replacement of the wild type RRLA↓ motif with RRRR↓ does switch LCMV dependence from SKI-1/S1P to Furin. Therefore, it seems that arenaviruses are not prompted to use other members of the PCs family. The reason(s) for this selectivity of SKI-1/S1P has not yet been elucidated.
3. Discussion
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
List of Abbreviations
References
- Dobson, A.P.; Carper, E.R. Infectious Diseases and Human Population History: Throughout history the establishment of disease has been a side effect of the growth of civilization. Bioscience 1996, 46, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Morens, D.M.; Folkers, G.K.; Fauci, A.S. Emerging infections: A perpetual challenge. Lancet Infect. Dis. 2008, 8, 710–719. [Google Scholar] [CrossRef]
- Cyranoski, D. Profile of a killer: The complex biology powering the coronavirus pandemic. Nature 2020, 581, 22–26. [Google Scholar] [CrossRef]
- Singh, R.; Kang, A.; Luo, X.; Jeyanathan, M.; Gillgrass, A.; Afkhami, S.; Xing, Z. COVID-19: Current knowledge in clinical features, immunological responses, and vaccine development. FASEB J. 2021, 35, e21409. [Google Scholar] [CrossRef] [PubMed]
- Funk, C.D.; Laferrière, C.; Ardakani, A. Target Product Profile Analysis of COVID-19 Vaccines in Phase III Clinical Trials and Beyond: An Early 2021 Perspective. Viruses 2021, 13, 418. [Google Scholar] [CrossRef]
- Morens, D.M.; Fauci, A.S. Emerging Pandemic Diseases: How We Got to COVID-19. Cell 2020, 182, 1077–1092. [Google Scholar] [CrossRef] [PubMed]
- Owusu, I.A.; Quaye, O.; Passalacqua, K.D.; Wobus, C.E. Egress of non-enveloped enteric RNA viruses. J. Gen. Virol. 2021, 102, 001557. [Google Scholar] [CrossRef]
- Leroy, H.; Han, M.; Woottum, M.; Bracq, L.; Bouchet, J.; Xie, M.; Benichou, S. Virus-Mediated Cell-Cell Fusion. Int. J. Mol. Sci. 2020, 21, 9644. [Google Scholar] [CrossRef]
- Seidah, N.G.; Prat, A. The biology and therapeutic targeting of the proprotein convertases. Nat. Rev. Drug Discov. 2012, 11, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Seidah, N.G.; Chretien, M. Proprotein and prohormone convertases: A family of subtilases generating diverse bioactive polypeptides. Brain Res. 1999, 848, 45–62. [Google Scholar] [CrossRef]
- Seidah, N.G.; Mowla, S.J.; Hamelin, J.; Mamarbachi, A.M.; Benjannet, S.; Toure, B.B.; Basak, A.; Munzer, J.S.; Marcinkiewicz, J.; Zhong, M.; et al. Mammalian subtilisin/kexin isozyme SKI-1: A widely expressed proprotein convertase with a unique cleavage specificity and cellular localization. Proc. Natl. Acad. Sci. USA 1999, 96, 1321–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espenshade, P.J.; Cheng, D.; Goldstein, J.L.; Brown, M.S. Autocatalytic processing of site-1 protease removes propeptide and permits cleavage of sterol regulatory element-binding proteins. J. Biol. Chem. 1999, 274, 22795–22804. [Google Scholar] [CrossRef] [Green Version]
- Pullikotil, P.; Vincent, M.; Nichol, S.T.; Seidah, N.G. Development of protein-based inhibitors of the proprotein of convertase SKI-1/S1P: Processing of SREBP-2, ATF6, and a viral glycoprotein. J. Biol. Chem. 2004, 279, 17338–17347. [Google Scholar] [CrossRef] [PubMed]
- Pasquato, A.; Pullikotil, P.; Asselin, M.C.; Vacatello, M.; Paolillo, L.; Ghezzo, F.; Basso, F.; Di Bello, C.; Dettin, M.; Seidah, N.G. The Proprotein Convertase SKI-1/S1P: In vitro analysis of lassa virus glycoprotein-derived substrates and ex vivo validation of irreversible peptide inhibitors. J. Biol. Chem. 2006, 281, 23471–23481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidah, N.G. Proprotein Convertases SKI-1/S1P and PCSK9. In Handbook of the Biologically Active Peptides; Minamino, N., Kastin, A., Eds.; Academic Press: Cambridge, MA, USA, 2012; pp. 1–8. [Google Scholar]
- Seidah, N.G.; Benjannet, S.; Wickham, L.; Marcinkiewicz, J.; Jasmin, S.B.; Stifani, S.; Basak, A.; Prat, A.; Chretien, M. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): Liver regeneration and neuronal differentiation. Proc. Natl. Acad. Sci. USA 2003, 100, 928–933. [Google Scholar] [CrossRef] [Green Version]
- Seidah, N.G.; Awan, Z.; Chretien, M.; Mbikay, M. PCSK9: A Key Modulator of Cardiovascular Health. Circ. Res. 2014, 114, 1022–1036. [Google Scholar] [CrossRef]
- Seidah, N.G.; Abifadel, M.; Prost, S.; Boileau, C.; Prat, A. The Proprotein Convertases in Hypercholesterolemia and Cardiovascular Diseases: Emphasis on Proprotein Convertase Subtilisin/Kexin 9. Pharmacol. Rev. 2017, 69, 33–52. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Bao, X.; Hu, M.; Chang, H.; Jiao, M.; Cheng, J.; Xie, L.; Huang, Q.; Li, F.; Li, C.Y. Inhibition of PCSK9 potentiates immune checkpoint therapy for cancer. Nature 2020, 588, 693–698. [Google Scholar] [CrossRef]
- Thomas, G. Furin at the cutting edge: From protein traffic to embryogenesis and disease. Nat. Rev. Mol. Cell Biol. 2002, 3, 753–766. [Google Scholar] [CrossRef] [Green Version]
- Izaguirre, G. The Proteolytic Regulation of Virus Cell Entry by Furin and Other Proprotein Convertases. Viruses 2019, 11, 837. [Google Scholar] [CrossRef] [Green Version]
- Van de Ven, W.J.; Creemers, J.W.; Roebroek, A.J. Furin: The prototype mammalian subtilisin-like proprotein-processing enzyme. Endoproteolytic cleavage at paired basic residues of proproteins of the eukaryotic secretory pathway. Enzyme 1991, 45, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Moulard, M.; Decroly, E. Maturation of HIV envelope glycoprotein precursors by cellular endoproteases. Biochim. Biophys. Acta 2000, 1469, 121–132. [Google Scholar] [CrossRef]
- Millet, J.K.; Whittaker, G.R. Host cell proteases: Critical determinants of coronavirus tropism and pathogenesis. Virus Res. 2015, 202, 120–134. [Google Scholar] [CrossRef]
- Braun, E.; Sauter, D. Furin-mediated protein processing in infectious diseases and cancer. Clin. Transl. Immunol. 2019, 8, e1073. [Google Scholar] [CrossRef] [Green Version]
- Le Coupanec, A.; Desforges, M.; Meessen-Pinard, M.; Dube, M.; Day, R.; Seidah, N.G.; Talbot, P.J. Cleavage of a Neuroinvasive Human Respiratory Virus Spike Glycoprotein by Proprotein Convertases Modulates Neurovirulence and Virus Spread within the Central Nervous System. PLoS Pathog. 2015, 11, e1005261. [Google Scholar] [CrossRef]
- Tang, T.; Bidon, M.; Jaimes, J.A.; Whittaker, G.R.; Daniel, S. Coronavirus membrane fusion mechanism offers as a potential target for antiviral development. Antivir. Res. 2020, 178, 104792. [Google Scholar] [CrossRef]
- Seidah, N.G. The proprotein convertases, 20 years later. Methods Mol. Biol. 2011, 768, 23–57. [Google Scholar]
- Hallenberger, S.; Bosch, V.; Angliker, H.; Shaw, E.; Klenk, H.D.; Garten, W. Inhibition of furin-mediated cleavage activation of HIV-1 glycoprotein gp160. Nature 1992, 360, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Stieneke-Grober, A.; Vey, M.; Angliker, H.; Shaw, E.; Thomas, G.; Roberts, C.; Klenk, H.D.; Garten, W. Influenza virus hemagglutinin with multibasic cleavage site is activated by furin, a subtilisin-like endoprotease. EMBO J. 1992, 11, 2407–2414. [Google Scholar] [CrossRef]
- Li, Z.; Sergel, T.; Razvi, E.; Morrison, T. Effect of cleavage mutants on syncytium formation directed by the wild-type fusion protein of Newcastle disease virus. J. Virol. 1998, 72, 3789–3795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, J.A.; Molloy, S.S.; Thomas, G.; Sakaguchi, T.; Yoshida, T.; Chambers, T.M.; Kawaoka, Y. Sequence specificity of furin, a proprotein-processing endoprotease, for the hemagglutinin of a virulent avian influenza virus. J. Virol. 1994, 68, 1213–1218. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Watanabe, S.; Ito, T.; Goto, H.; Wells, K.; McGregor, M.; Cooley, A.J.; Kawaoka, Y. Biological heterogeneity, including systemic replication in mice, of H5N1 influenza A virus isolates from humans in Hong Kong. J. Virol. 1999, 73, 3184–3189. [Google Scholar] [CrossRef] [Green Version]
- Claas, E.C.; Osterhaus, A.D.; van Beek, R.; de Jong, J.C.; Rimmelzwaan, G.F.; Senne, D.A.; Krauss, S.; Shortridge, K.F.; Webster, R.G. Human influenza A H5N1 virus related to a highly pathogenic avian influenza virus. Lancet 1998, 351, 472–477. [Google Scholar] [CrossRef]
- Basak, A.; Zhong, M.; Munzer, J.S.; Chretien, M.; Seidah, N.G. Implication of the proprotein convertases furin, PC5 and PC7 in the cleavage of surface glycoproteins of Hong Kong, Ebola and respiratory syncytial viruses: A comparative analysis with fluorogenic peptides. Biochem. J. 2001, 353, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, E.; Matrosovich, T.; Beyerle, M.; Klenk, H.D.; Garten, W.; Matrosovich, M. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J. Virol. 2006, 80, 9896–9898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murza, A.; Dion, S.P.; Boudreault, P.L.; Désilets, A.; Leduc, R.; Marsault, É. Inhibitors of type II transmembrane serine proteases in the treatment of diseases of the respiratory tract—A review of patent literature. Expert Opin. Ther. Pat. 2020, 30, 807–824. [Google Scholar] [CrossRef] [PubMed]
- Sachan, V.; Lodge, R.; Mihara, K.; Hamelin, J.; Power, C.; Gelman, B.B.; Hollenberg, M.D.; Cohen, É.A.; Seidah, N.G. HIV-induced neuroinflammation: Impact of PAR1 and PAR2 processing by Furin. Cell Death Differ. 2019, 26, 1942–1954. [Google Scholar] [CrossRef] [PubMed]
- Ohuchi, M.; Orlich, M.; Ohuchi, R.; Simpson, B.E.; Garten, W.; Klenk, H.D.; Rott, R. Mutations at the cleavage site of the hemagglutinin after the pathogenicity of influenza virus A/chick/Penn/83 (H5N2). Virology 1989, 168, 274–280. [Google Scholar] [CrossRef]
- Gu, M.; Rappaport, J.; Leppla, S.H. Furin is important but not essential for the proteolytic maturation of gp160 of HIV-1. FEBS Lett. 1995, 365, 95–97. [Google Scholar] [CrossRef] [Green Version]
- Aerts, L.; Hamelin, M.E.; Rhaume, C.; Lavigne, S.; Couture, C.; Kim, W.; Susan-Resiga, D.; Prat, A.; Seidah, N.G.; Vergnolle, N.; et al. Modulation of protease activated receptor 1 influences human metapneumovirus disease severity in a mouse model. PLoS ONE 2013, 8, e72529. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Zekas, E.; Lodge, R.; Susan-Resiga, D.; Marcinkiewicz, E.; Essalmani, R.; Mihara, K.; Ramachandran, R.; Asahchop, E.; Gelman, B.; et al. Neuroinflammation-Induced Interactions between Protease-Activated Receptor 1 and Proprotein Convertases in HIV-Associated Neurocognitive Disorder. Mol. Cell Biol. 2015, 35, 3684–3700. [Google Scholar] [CrossRef] [Green Version]
- Braun, E.; Hotter, D.; Koepke, L.; Zech, F.; Groß, R.; Sparrer, K.M.J.; Müller, J.A.; Pfaller, C.K.; Heusinger, E.; Wombacher, R.; et al. Guanylate-Binding Proteins 2 and 5 Exert Broad Antiviral Activity by Inhibiting Furin-Mediated Processing of Viral Envelope Proteins. Cell Rep. 2019, 27, 2092–2104.e10. [Google Scholar] [CrossRef] [Green Version]
- Volchkov, V.E.; Feldmann, H.; Volchkova, V.A.; Klenk, H.D. Processing of the Ebola virus glycoprotein by the proprotein convertase furin. Proc. Natl. Acad. Sci. USA 1998, 95, 5762–5767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, G.; Feldmann, H.; Watanabe, S.; Lukashevich, I.; Kawaoka, Y. Reverse genetics demonstrates that proteolytic processing of the Ebola virus glycoprotein is not essential for replication in cell culture. J. Virol. 2002, 76, 406–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, G.; Geisbert, T.W.; Ebihara, H.; Geisbert, J.B.; Daddario-DiCaprio, K.M.; Feldmann, H.; Kawaoka, Y. Proteolytic processing of the Ebola virus glycoprotein is not critical for Ebola virus replication in nonhuman primates. J. Virol. 2007, 81, 2995–2998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Wang, Y.; Frabutt, D.A.; Zhang, X.; Yao, X.; Hu, D.; Zhang, Z.; Liu, C.; Zheng, S.; Xiang, S.H.; et al. Mechanistic understanding of N-glycosylation in Ebola virus glycoprotein maturation and function. J. Biol. Chem. 2017, 292, 5860–5870. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Li, S.; Zhang, X.; Khan, I.; Ahmad, I.; Zhou, Y.; Li, S.; Shi, J.; Wang, Y.; Zheng, Y.H. MARCH8 Inhibits Ebola Virus Glycoprotein, Human Immunodeficiency Virus Type 1 Envelope Glycoprotein, and Avian Influenza Virus H5N1 Hemagglutinin Maturation. mBio 2020, 11, e01882-20. [Google Scholar] [CrossRef]
- Stadler, K.; Allison, S.L.; Schalich, J.; Heinz, F.X. Proteolytic activation of tick-borne encephalitis virus by furin. J. Virol. 1997, 71, 8475–8481. [Google Scholar] [CrossRef] [Green Version]
- Zybert, I.A.; van der Ende-Metselaar, H.; Wilschut, J.; Smit, J.M. Functional importance of dengue virus maturation: Infectious properties of immature virions. J. Gen. Virol. 2008, 89, 3047–3051. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Voorham, J.M.; Rodenhuis-Zybert, I.A.; Ayala Nuñez, N.V.; Colpitts, T.M.; van der Ende-Metselaar, H.; Fikrig, E.; Diamond, M.S.; Wilschut, J.; Smit, J.M. Antibodies against the envelope glycoprotein promote infectivity of immature dengue virus serotype 2. PLoS ONE 2012, 7, e29957. [Google Scholar] [CrossRef] [Green Version]
- Rodenhuis-Zybert, I.A.; van der Schaar, H.M.; da Silva Voorham, J.M.; van der Ende-Metselaar, H.; Lei, H.Y.; Wilschut, J.; Smit, J.M. Immature dengue virus: A veiled pathogen? PLoS Pathog. 2010, 6, e1000718. [Google Scholar] [CrossRef] [Green Version]
- VanBlargan, L.A.; Errico, J.M.; Kafai, N.M.; Burgomaster, K.E.; Jethva, P.N.; Broeckel, R.M.; Meade-White, K.; Nelson, C.A.; Himansu, S.; Wang, D.; et al. Broadly neutralizing monoclonal antibodies protect against multiple tick-borne flaviviruses. J. Exp. Med. 2021, 218, e20210174. [Google Scholar] [CrossRef] [PubMed]
- Ozden, S.; Lucas-Hourani, M.; Ceccaldi, P.E.; Basak, A.; Valentine, M.; Benjannet, S.; Hamelin, J.; Jacob, Y.; Mamchaoui, K.; Mouly, V.; et al. Inhibition of Chikungunya Virus Infection in Cultured Human Muscle Cells by Furin Inhibitors: Impairment of the maturation of the E2 surface glycoprotein. J. Biol. Chem. 2008, 283, 21899–21908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kropp, K.A.; Srivaratharajan, S.; Ritter, B.; Yu, P.; Krooss, S.; Polten, F.; Pich, A.; Alcami, A.; Viejo-Borbolla, A. Identification of the Cleavage Domain within Glycoprotein G of Herpes Simplex Virus Type 2. Viruses 2020, 12, 1428. [Google Scholar] [CrossRef] [PubMed]
- Oliver, S.L.; Sommer, M.; Zerboni, L.; Rajamani, J.; Grose, C.; Arvin, A.M. Mutagenesis of varicella-zoster virus glycoprotein B: Putative fusion loop residues are essential for viral replication, and the furin cleavage motif contributes to pathogenesis in skin tissue in vivo. J. Virol. 2009, 83, 7495–7506. [Google Scholar] [CrossRef] [Green Version]
- Emerman, M.; Malim, M.H. HIV-1 regulatory/accessory genes: Keys to unraveling viral and host cell biology. Science 1998, 280, 1880–1884. [Google Scholar] [CrossRef]
- Wallet, C.; Rohr, O.; Schwartz, C. Evolution of a concept: From accessory protein to key virulence factor, the case of HIV-1 Vpr. Biochem. Pharmacol. 2020, 180, 114128. [Google Scholar] [CrossRef]
- Mayer, G.; Hamelin, J.; Asselin, M.C.; Pasquato, A.; Marcinkiewicz, E.; Tang, M.; Tabibzadeh, S.; Seidah, N.G. The regulated cell surface zymogen activation of the proprotein convertase PC5A directs the processing of its secretory substrates. J. Biol. Chem. 2008, 283, 2373–2384. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Chen, G.; Richard, J.; Rougeau, N.; Li, H.; Seidah, N.G.; Cohen, E.A. Cell-surface processing of extracellular human immunodeficiency virus type 1 Vpr by proprotein convertases. Virology 2008, 372, 384–397. [Google Scholar] [CrossRef] [Green Version]
- Bosch, B.J.; van der, Z.R.; de Haan, C.A.; Rottier, P.J. The coronavirus spike protein is a class I virus fusion protein: Structural and functional characterization of the fusion core complex. J. Virol. 2003, 77, 8801–8811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Wit, E.; van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, G.; Hu, Y.; Wang, Q.; Qi, J.; Gao, F.; Li, Y.; Zhang, Y.; Zhang, W.; Yuan, Y.; Bao, J.; et al. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature 2013, 500, 227–231. [Google Scholar] [CrossRef] [Green Version]
- Belouzard, S.; Madu, I.; Whittaker, G.R. Elastase-mediated activation of the severe acute respiratory syndrome coronavirus spike protein at discrete sites within the S2 domain. J. Biol. Chem. 2010, 285, 22758–22763. [Google Scholar] [CrossRef] [Green Version]
- Belouzard, S.; Millet, J.K.; Licitra, B.N.; Whittaker, G.R. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 2012, 4, 1011–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirato, K.; Kawase, M.; Matsuyama, S. Middle East respiratory syndrome coronavirus infection mediated by the transmembrane serine protease TMPRSS2. J. Virol. 2013, 87, 12552–12561. [Google Scholar] [CrossRef] [Green Version]
- Kleine-Weber, H.; Elzayat, M.T.; Hoffmann, M.; Pöhlmann, S. Functional analysis of potential cleavage sites in the MERS-coronavirus spike protein. Sci. Rep. 2018, 8, 16597. [Google Scholar] [CrossRef] [PubMed]
- Hörnich, B.F.; Großkopf, A.K.; Schlagowski, S.; Tenbusch, M.; Kleine-Weber, H.; Neipel, F.; Stahl-Hennig, C.; Hahn, A.S. SARS-CoV-2 and SARS-CoV spike-mediated cell-cell fusion differ in the requirements for receptor expression and proteolytic activation. J. Virol. 2021, 95, e00002-21. [Google Scholar] [CrossRef] [PubMed]
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 2020, 176, 104742. [Google Scholar] [CrossRef]
- Bollavaram, K.; Leeman, T.H.; Lee, M.W.; Kulkarni, A.; Upshaw, S.G.; Yang, J.; Song, H.; Platt, M.O. Multiple Sites on SARS-CoV-2 Spike Protein are Susceptible to Proteolysis by Cathepsins B, K, L, S, and V. Protein Sci. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kawase, M.; Shirato, K.; van der Hoek, L.; Taguchi, F.; Matsuyama, S. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J. Virol. 2012, 86, 6537–6545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimishakavi, S.; Raymond, W.W.; Gruenert, D.C.; Caughey, G.H. Divergent Inhibitor Susceptibility among Airway Lumen-Accessible Tryptic Proteases. PLoS ONE 2015, 10, e0141169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essalmani, R.; Jain, J.; Susan-Resiga, D.; Andréo, U.; Evagelidis, A.; Derbali, R.M.; Huynh, D.N.; Dallaire, F.; Laporte, M.; Delpal, A.; et al. Furin cleaves SARS-CoV-2 spike-glycoprotein at S1/S2 and S2′ for viral fusion/entry: Indirect role of TMPRSS2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Raghuvamsi, P.V.; Tulsian, N.K.; Samsudin, F.; Qian, X.; Purushotorman, K.; Yue, G.; Kozma, M.M.; Hwa, W.Y.; Lescar, J.; Bond, P.J.; et al. SARS-CoV-2 S protein:ACE2 interaction reveals novel allosteric targets. eLife 2021, 10, e63646. [Google Scholar] [CrossRef]
- Tang, T.; Jaimes, J.A.; Bidon, M.K.; Straus, M.R.; Daniel, S.; Whittaker, G.R. Proteolytic Activation of SARS-CoV-2 Spike at the S1/S2 Boundary: Potential Role of Proteases beyond Furin. ACS Infect Dis. 2021, 7, 264–272. [Google Scholar] [CrossRef]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pöhlmann, S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef] [Green Version]
- Kishimoto, M.; Uemura, K.; Sanaki, T.; Sato, A.; Hall, W.W.; Kariwa, H.; Orba, Y.; Sawa, H.; Sasaki, M. TMPRSS11D and TMPRSS13 Activate the SARS-CoV-2 Spike Protein. Viruses 2021, 13, 384. [Google Scholar] [CrossRef]
- Vincent, M.J.; Bergeron, E.; Benjannet, S.; Erickson, B.R.; Rollin, P.E.; Ksiazek, T.G.; Seidah, N.G.; Nichol, S.T. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol. J. 2005, 2, 69. [Google Scholar] [CrossRef] [Green Version]
- Hache, G.; Rolain, J.M.; Gautret, P.; Deharo, J.C.; Brouqui, P.; Raoult, D.; Honoré, S. Combination of Hydroxychloroquine Plus Azithromycin As Potential Treatment for COVID-19 Patients: Safety Profile, Drug Interactions, and Management of Toxicity. Microb. Drug Resist. 2021, 27, 281–290. [Google Scholar] [CrossRef]
- Maisonnasse, P.; Guedj, J.; Contreras, V.; Behillil, S.; Solas, C.; Marlin, R.; Naninck, T.; Pizzorno, A.; Lemaitre, J.; Gonçalves, A.; et al. Hydroxychloroquine use against SARS-CoV-2 infection in non-human primates. Nature 2020, 585, 584–587. [Google Scholar] [CrossRef]
- Leung, K.; Shum, M.H.; Leung, G.M.; Lam, T.T.; Wu, J.T. Early transmissibility assessment of the N501Y mutant strains of SARS-CoV-2 in the United Kingdom, October to November 2020. Eurosurveillance 2021, 26, 2002106. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv 2020. [Google Scholar] [CrossRef]
- Fujino, T.; Nomoto, H.; Kutsuna, S.; Ujiie, M.; Suzuki, T.; Sato, R.; Fujimoto, T.; Kuroda, M.; Wakita, T.; Ohmagari, N. Novel SARS-CoV-2 Variant Identified in Travelers from Brazil to Japan. Emerg. Infect. Dis. 2021, 27, 1243. [Google Scholar] [CrossRef]
- Zahradník, J.; Marciano, S.; Shemesh, M.; Zoler, E.; Chiaravalli, J.; Meyer, B.; Dym, O.; Elad, N.; Schreiber, G. SARS-CoV-2 RBD in vitro evolution follows contagious mutation spread, yet generates an able infection inhibitor. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wang, P.; Nair, M.S.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; et al. Antibody Resistance of SARS-CoV-2 Variants B.1.351 and B.1.1.7. Nature 2021, 593, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.E.; Zhang, X.; Case, J.B.; Winkler, E.S.; Liu, Y.; VanBlargan, L.A.; Liu, J.; Errico, J.M.; Xie, X.; Suryadevara, N.; et al. Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat. Med. 2021, 27, 717–726. [Google Scholar] [CrossRef]
- Yang, C.; Huang, Y.; Liu, S. Therapeutic Development in COVID-19. Adv. Exp. Med. Biol. 2021, 1318, 435–448. [Google Scholar] [PubMed]
- Benjannet, S.; Rhainds, D.; Essalmani, R.; Mayne, J.; Wickham, L.; Jin, W.; Asselin, M.C.; Hamelin, J.; Varret, M.; Allard, D.; et al. NARC-1/PCSK9 and its natural mutants: Zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J. Biol. Chem. 2004, 279, 48865–48875. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Tumanut, C.; Gavigan, J.A.; Huang, W.J.; Hampton, E.N.; Tumanut, R.; Suen, K.F.; Trauger, J.W.; Spraggon, G.; Lesley, S.A.; et al. Secreted PCSK9 promotes LDL receptor degradation independently of proteolytic activity. Biochem. J. 2007, 406, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Abifadel, M.; Varret, M.; Rabes, J.P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef]
- Cohen, J.; Pertsemlidis, A.; Kotowski, I.K.; Graham, R.; Garcia, C.K.; Hobbs, H.H. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 2005, 37, 161. [Google Scholar] [CrossRef]
- Maxwell, K.N.; Breslow, J.L. Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc. Natl. Acad. Sci. USA 2004, 101, 7100–7105. [Google Scholar] [CrossRef] [Green Version]
- Park, S.W.; Moon, Y.A.; Horton, J.D. Post-transcriptional regulation of low density lipoprotein receptor protein by proprotein convertase subtilisin/kexin type 9a in mouse liver. J. Biol. Chem. 2004, 279, 50630–50638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syed, G.H.; Tang, H.; Khan, M.; Hassanein, T.; Liu, J.; Siddiqui, A. Hepatitis C virus stimulates low-density lipoprotein receptor expression to facilitate viral propagation. J. Virol. 2014, 88, 2519–2529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Liu, Q. Hepatitis C virus regulates proprotein convertase subtilisin/kexin type 9 promoter activity. Biochem. Biophys. Res. Commun. 2018, 496, 1229–1235. [Google Scholar] [CrossRef]
- Ramanathan, A.; Gusarova, V.; Stahl, N.; Gurnett-Bander, A.; Kyratsous, C.A. Alirocumab, a Therapeutic Human Antibody to PCSK9, Does Not Affect CD81 Levels or Hepatitis C Virus Entry and Replication into Hepatocytes. PLoS ONE 2016, 11, e0154498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridge, S.H.; Sheridan, D.A.; Felmlee, D.J.; Crossey, M.M.; Fenwick, F.I.; Lanyon, C.V.; Dubuc, G.; Seidah, N.G.; Davignon, J.; Thomas, H.C.; et al. PCSK9, apolipoprotein E and lipoviral particles in chronic hepatitis C genotype 3: Evidence for genotype-specific regulation of lipoprotein metabolism. J. Hepatol. 2014, 62, 763–770. [Google Scholar] [CrossRef] [Green Version]
- Fasolato, S.; Pigozzo, S.; Pontisso, P.; Angeli, P.; Ruscica, M.; Savarino, E.; De Martin, S.; Lupo, M.G.; Ferri, N. PCSK9 Levels Are Raised in Chronic HCV Patients with Hepatocellular Carcinoma. J. Clin. Med. 2020, 9, 3134. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Junjhon, J.; Lausumpao, M.; Supasa, S.; Noisakran, S.; Songjaeng, A.; Saraithong, P.; Chaichoun, K.; Utaipat, U.; Keelapang, P.; Kanjanahaluethai, A.; et al. Differential modulation of prM cleavage, extracellular particle distribution, and virus infectivity by conserved residues at nonfurin consensus positions of the dengue virus pr-M junction. J. Virol. 2008, 82, 10776–10791. [Google Scholar] [CrossRef] [Green Version]
- Rana, J.; Slon Campos, J.L.; Poggianella, M.; Burrone, O.R. Dengue virus capsid anchor modulates the efficiency of polyprotein processing and assembly of viral particles. J. Gen. Virol. 2019, 100, 1663–1673. [Google Scholar] [CrossRef]
- Gan, E.S.; Tan, H.C.; Le, D.H.T.; Huynh, T.T.; Wills, B.; Seidah, N.G.; Ooi, E.E.; Yacoub, S. Dengue virus induces PCSK9 expression to alter antiviral responses and disease outcomes. J. Clin. Investig. 2020, 130, 5223–5234. [Google Scholar] [CrossRef] [PubMed]
- Sakai, J.; Rawson, R.B.; Espenshade, P.J.; Cheng, D.; Seegmiller, A.C.; Goldstein, J.L.; Brown, M.S. Molecular identification of the sterol-regulated luminal protease that cleaves SREBPs and controls lipid composition of animal cells. Mol. Cell 1998, 2, 505–514. [Google Scholar] [CrossRef]
- Da Palma, J.R.; Burri, D.J.; Oppliger, J.; Salamina, M.; Cendron, L.; de Laureto, P.P.; Seidah, N.G.; Kunz, S.; Pasquato, A. Zymogen activation and subcellular activity of subtilisin kexin isozyme 1/site 1 protease. J. Biol. Chem. 2014, 289, 35743–35756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tagliabracci, V.S.; Wiley, S.E.; Guo, X.; Kinch, L.N.; Durrant, E.; Wen, J.; Xiao, J.; Cui, J.; Nguyen, K.B.; Engel, J.L.; et al. A Single Kinase Generates the Majority of the Secreted Phosphoproteome. Cell 2015, 161, 1619–1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marschner, K.; Kollmann, K.; Schweizer, M.; Braulke, T.; Pohl, S. A key enzyme in the biogenesis of lysosomes is a protease that regulates cholesterol metabolism. Science 2011, 333, 87–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, T.; Suzuki-Nakagawa, C.; Watanabe, A.; Asami, E.; Matsumoto, M.; Nakano, M.; Ebihara, A.; Uddin, M.N.; Suzuki, F. Site-1 protease is required for the generation of soluble (pro)renin receptor. J. Biochem. 2017, 161, 369–379. [Google Scholar] [CrossRef]
- Kondo, Y.; Fu, J.; Wang, H.; Hoover, C.; McDaniel, J.M.; Steet, R.; Patra, D.; Song, J.; Pollard, L.; Cathey, S.; et al. Site-1 protease deficiency causes human skeletal dysplasia due to defective inter-organelle protein trafficking. JCI Insight 2018, 3, e121596. [Google Scholar] [CrossRef]
- Meyer, R.; Elbracht, M.; Opladen, T.; Eggermann, T. Patient with an autosomal-recessive MBTPS1-linked phenotype and clinical features of Silver-Russell syndrome. Am. J. Med. Genet. A 2020, 182, 2727–2730. [Google Scholar] [CrossRef]
- Lenz, O.; ter Meulen, J.; Klenk, H.D.; Seidah, N.G.; Garten, W. The Lassa virus glycoprotein precursor GP-C is proteolytically processed by subtilase SKI-1/S1P. Proc. Natl. Acad. Sci. USA 2001, 98, 12701–12705. [Google Scholar] [CrossRef] [Green Version]
- Burri, D.J.; Pasqual, G.; Rochat, C.; Seidah, N.G.; Pasquato, A.; Kunz, S. Molecular characterization of the processing of arenavirus envelope glycoprotein precursors by subtilisin kexin isozyme-1/site-1 protease. J. Virol. 2012, 86, 4935–4946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burri, D.J.; da Palma, J.R.; Seidah, N.G.; Zanotti, G.; Cendron, L.; Pasquato, A.; Kunz, S. Differential recognition of Old World and New World arenavirus envelope glycoproteins by subtilisin kexin isozyme 1 (SKI-1)/site 1 protease (S1P). J. Virol. 2013, 87, 6406–6414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormick, J.B.; Webb, P.A.; Krebs, J.W.; Johnson, K.M.; Smith, E.S. A prospective study of the epidemiology and ecology of Lassa fever. J. Infect. Dis. 1987, 155, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Da Palma, J.R.; Cendron, L.; Seidah, N.G.; Pasquato, A.; Kunz, S. Mechanism of Folding and Activation of Subtilisin Kexin Isozyme-1 (SKI-1)/Site-1 Protease (S1P). J. Biol. Chem. 2016, 291, 2055–2066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flatz, L.; Rieger, T.; Merkler, D.; Bergthaler, A.; Regen, T.; Schedensack, M.; Bestmann, L.; Verschoor, A.; Kreutzfeldt, M.; Brück, W.; et al. T cell-dependence of Lassa fever pathogenesis. PLoS Pathog. 2010, 6, e1000836. [Google Scholar] [CrossRef] [Green Version]
- Seidah, N.G.; Prat, A.; Pirillo, A.; Catapano, A.L.; Norata, G.D. Novel strategies to target proprotein convertase subtilisin kexin 9: Beyond monoclonal antibodies. Cardiovasc. Res. 2019, 115, 510–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatch, S.; Mathew, A.; Rothman, A. Dengue vaccine: Opportunities and challenges. IDrugs 2008, 11, 42–45. [Google Scholar]
- Beyer, W.R.; Popplau, D.; Garten, W.; Von Laer, D.; Lenz, O. Endoproteolytic Processing of the Lymphocytic Choriomeningitis Virus Glycoprotein by the Subtilase SKI-1/S1P. J. Virol. 2003, 77, 2866–2872. [Google Scholar] [CrossRef] [Green Version]
- Bederka, L.H.; Bonhomme, C.J.; Ling, E.L.; Buchmeier, M.J. Arenavirus stable signal peptide is the keystone subunit for glycoprotein complex organization. MBio 2014, 5, e02063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunz, S.; Edelmann, K.H.; de la Torre, J.C.; Gorney, R.; Oldstone, M.B. Mechanisms for lymphocytic choriomeningitis virus glycoprotein cleavage, transport, and incorporation into virions. Virology 2003, 314, 168–178. [Google Scholar] [CrossRef] [Green Version]
- Wright, K.E.; Spiro, R.C.; Burns, J.W.; Buchmeier, M.J. Post-translational processing of the glycoproteins of lymphocytic choriomeningitis virus. Virology 1990, 177, 175–183. [Google Scholar] [CrossRef]
- Briese, T.; Paweska, J.T.; McMullan, L.K.; Hutchison, S.K.; Street, C.; Palacios, G.; Khristova, M.L.; Weyer, J.; Swanepoel, R.; Egholm, M.; et al. Genetic detection and characterization of Lujo virus, a new hemorrhagic fever-associated arenavirus from southern Africa. PLoS Pathog. 2009, 5, e1000455. [Google Scholar] [CrossRef] [Green Version]
- Oppliger, J.; da Palma, J.R.; Burri, D.J.; Bergeron, E.; Khatib, A.M.; Spiropoulou, C.F.; Pasquato, A.; Kunz, S. A Molecular Sensor to Characterize Arenavirus Envelope Glycoprotein Cleavage by Subtilisin Kexin Isozyme 1/Site 1 Protease. J. Virol. 2016, 90, 705–714. [Google Scholar] [CrossRef] [Green Version]
- Grant, A.; Seregin, A.; Huang, C.; Kolokoltsova, O.; Brasier, A.; Peters, C.; Paessler, S. Junín virus pathogenesis and virus replication. Viruses 2012, 4, 2317–2339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Candurra, N.A.; Damonte, E.B. Effect of inhibitors of the intracellular exocytic pathway on glycoprotein processing and maturation of Junin virus. Arch. Virol. 1997, 142, 2179–2193. [Google Scholar] [CrossRef] [PubMed]
- Toure, B.B.; Munzer, J.S.; Basak, A.; Benjannet, S.; Rochemont, J.; Lazure, C.; Chretien, M.; Seidah, N.G. Biosynthesis and enzymatic characterization of human SKI-1/S1P and the processing of its inhibitory prosegment. J. Biol. Chem. 2000, 275, 2349–2358. [Google Scholar] [CrossRef] [Green Version]
- Pasquato, A.; Burri, D.J.; Traba, E.G.; Hanna-El-Daher, L.; Seidah, N.G.; Kunz, S. Arenavirus envelope glycoproteins mimic autoprocessing sites of the cellular proprotein convertase subtilisin kexin isozyme-1/site-1 protease. Virology 2011, 417, 18–26. [Google Scholar] [CrossRef] [Green Version]
- Pasqual, G.; Burri, D.J.; Pasquato, A.; de la Torre, J.C.; Kunz, S. Role of the host cell’s unfolded protein response in arenavirus infection. J. Virol. 2011, 85, 1662–1670. [Google Scholar] [CrossRef] [Green Version]
- Manning, J.T.; Yun, N.E.; Seregin, A.V.; Koma, T.; Sattler, R.A.; Ezeomah, C.; Huang, C.; de la Torre, J.C.; Paessler, S. The Glycoprotein of the Live-Attenuated Junin Virus Vaccine Strain Induces Endoplasmic Reticulum Stress and Forms Aggregates prior to Degradation in the Lysosome. J. Virol. 2020, 94, e01693-19. [Google Scholar] [CrossRef] [PubMed]
- Popkin, D.L.; Teijaro, J.R.; Sullivan, B.M.; Urata, S.; Rutschmann, S.; de la Torre, J.C.; Kunz, S.; Beutler, B.; Oldstone, M. Hypomorphic mutation in the site-1 protease Mbtps1 endows resistance to persistent viral infection in a cell-specific manner. Cell Host Microbe 2011, 9, 212–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burri, D.J.; da Palma, J.R.; Kunz, S.; Pasquato, A. Envelope glycoprotein of arenaviruses. Viruses 2012, 4, 2162–2181. [Google Scholar] [CrossRef] [PubMed]
- Maisa, A.; Stroher, U.; Klenk, H.D.; Garten, W.; Strecker, T. Inhibition of Lassa virus glycoprotein cleavage and multicycle replication by site 1 protease-adapted α 1-antitrypsin variants. PLoS. Negl. Trop. Dis. 2009, 3, e446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basak, A.; Goswami, M.; Rajkumar, A.; Mitra, T.; Majumdar, S.; O’Reilly, P.; Bdour, H.M.; Trudeau, V.L.; Basak, A. Enediynyl peptides and iso-coumarinyl methyl sulfones as inhibitors of proprotein convertases PCSK8/SKI-1/S1P and PCSK4/PC4: Design, synthesis and biological evaluations. Bioorg. Med. Chem. Lett. 2015, 25, 2225–2237. [Google Scholar] [CrossRef]
- Rojek, J.M.; Pasqual, G.; Sanchez, A.B.; Nguyen, N.T.; de la Torre, J.C.; Kunz, S. Targeting the proteolytic processing of the viral glycoprotein precursor is a promising novel antiviral strategy against arenaviruses. J. Virol. 2010, 84, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Hay, B.A.; Abrams, B.; Zumbrunn, A.Y.; Valentine, J.J.; Warren, L.C.; Petras, S.F.; Shelly, L.D.; Xia, A.; Varghese, A.H.; Hawkins, J.L.; et al. Aminopyrrolidineamide inhibitors of site-1 protease. Bioorg. Med. Chem. Lett. 2007, 17, 4411–4414. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, J.L.; Robbins, M.D.; Warren, L.C.; Xia, D.; Petras, S.F.; Valentine, J.J.; Varghese, A.H.; Wang, I.K.; Subashi, T.A.; Shelly, L.D.; et al. Pharmacologic inhibition of site 1 protease activity inhibits sterol regulatory element-binding protein processing and reduces lipogenic enzyme gene expression and lipid synthesis in cultured cells and experimental animals. J. Pharmacol. Exp. Ther. 2008, 326, 801–808. [Google Scholar] [CrossRef] [Green Version]
- Urata, S.; Yun, N.; Pasquato, A.; Paessler, S.; Kunz, S.; de la Torre, J.C. Antiviral activity of a small-molecule inhibitor of arenavirus glycoprotein processing by the cellular site 1 protease. J. Virol. 2011, 85, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Pasquato, A.; Rochat, C.; Burri, D.J.; Pasqual, G.; la Torre, J.C.; Kunz, S. Evaluation of the anti-arenaviral activity of the subtilisin kexin isozyme-1/site-1 protease inhibitor PF-429242. Virology 2012, 423, 14–22. [Google Scholar] [CrossRef] [Green Version]
- Vincent, M.J.; Sanchez, A.J.; Erickson, B.R.; Basak, A.; Chretien, M.; Seidah, N.G.; Nichol, S.T. Crimean-Congo hemorrhagic fever virus glycoprotein proteolytic processing by subtilase SKI-1. J. Virol. 2003, 77, 8640–8649. [Google Scholar] [CrossRef] [Green Version]
- Bergeron, E.; Vincent, M.J.; Nichol, S.T. Crimean-Congo hemorrhagic fever virus glycoprotein processing by the endoprotease SKI-1/S1P is critical for virus infectivity. J. Virol. 2007, 81, 13271–13276. [Google Scholar] [CrossRef] [Green Version]
- Bergeron, É.; Zivcec, M.; Chakrabarti, A.K.; Nichol, S.T.; Albariño, C.G.; Spiropoulou, C.F. Recovery of Recombinant Crimean Congo Hemorrhagic Fever Virus Reveals a Function for Non-structural Glycoproteins Cleavage by Furin. PLoS Pathog. 2015, 11, e1004879. [Google Scholar] [CrossRef] [Green Version]
- Flint, M.; Chatterjee, P.; Lin, D.L.; McMullan, L.K.; Shrivastava-Ranjan, P.; Bergeron, É.; Lo, M.K.; Welch, S.R.; Nichol, S.T.; Tai, A.W.; et al. A genome-wide CRISPR screen identifies N-acetylglucosamine-1-phosphate transferase as a potential antiviral target for Ebola virus. Nat. Commun. 2019, 10, 285. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Chu, H.; Chan, J.F.; Ye, Z.W.; Wen, L.; Yan, B.; Lai, P.M.; Tee, K.M.; Huang, J.; Chen, D.; et al. SREBP-dependent lipidomic reprogramming as a broad-spectrum antiviral target. Nat. Commun. 2019, 10, 120. [Google Scholar] [CrossRef]
- Wang, R.; Simoneau, C.R.; Kulsuptrakul, J.; Bouhaddou, M.; Travisano, K.A.; Hayashi, J.M.; Carlson-Stevermer, J.; Zengel, J.R.; Richards, C.M.; Fozouni, P.; et al. Genetic Screens Identify Host Factors for SARS-CoV-2 and Common Cold Coronaviruses. Cell 2021, 184, 106–119.e14. [Google Scholar] [CrossRef]
- Dias, S.S.G.; Soares, V.C.; Ferreira, A.C.; Sacramento, C.Q.; Fintelman-Rodrigues, N.; Temerozo, J.R.; Teixeira, L.; Nunes da Silva, M.A.; Barreto, E.; Mattos, M.; et al. Lipid droplets fuel SARS-CoV-2 replication and production of inflammatory mediators. PLoS Pathog. 2020, 16, e1009127. [Google Scholar] [CrossRef]
- Lee, W.; Ahn, J.H.; Park, H.H.; Kim, H.N.; Kim, H.; Yoo, Y.; Shin, H.; Hong, K.S.; Jang, J.G.; Park, C.G.; et al. COVID-19-activated SREBP2 disturbs cholesterol biosynthesis and leads to cytokine storm. Signal Transduct. Target 2020, 5, 186. [Google Scholar] [CrossRef] [PubMed]
- Sohrabi, Y.; Reinecke, H.; Godfrey, R. Altered Cholesterol and Lipid Synthesis Mediates Hyperinflammation in COVID-19. Trends Endocrinol. Metab. 2021, 32, 132–134. [Google Scholar] [CrossRef] [PubMed]
- Hyrina, A.; Meng, F.; McArthur, S.J.; Eivemark, S.; Nabi, I.R.; Jean, F. Human Subtilisin Kexin Isozyme-1 (SKI-1)/Site-1 Protease (S1P) regulates cytoplasmic lipid droplet abundance: A potential target for indirect-acting anti-dengue virus agents. PLoS ONE 2017, 12, e0174483. [Google Scholar] [CrossRef] [PubMed]
- Blanchet, M.; Sureau, C.; Guevin, C.; Seidah, N.G.; Labonte, P. SKI-1/S1P inhibitor PF-429242 impairs the onset of HCV infection. Antivir. Res. 2015, 115, 94–104. [Google Scholar] [CrossRef]
- Olmstead, A.D.; Knecht, W.; Lazarov, I.; Dixit, S.B.; Jean, F. Human subtilase SKI-1/S1P is a master regulator of the HCV Lifecycle and a potential host cell target for developing indirect-acting antiviral agents. PLoS Pathog. 2012, 8, e1002468. [Google Scholar] [CrossRef] [Green Version]
- Merino-Ramos, T.; Jiménez de Oya, N.; Saiz, J.C.; Martín-Acebes, M.A. Antiviral Activity of Nordihydroguaiaretic Acid and Its Derivative Tetra-O-Methyl Nordihydroguaiaretic Acid against West Nile Virus and Zika Virus. Antimicrob. Agents Chemother. 2017, 61, e00376-17. [Google Scholar] [CrossRef] [Green Version]
- Uchida, L.; Urata, S.; Ulanday, G.E.; Takamatsu, Y.; Yasuda, J.; Morita, K.; Hayasaka, D. Suppressive Effects of the Site 1 Protease (S1P) Inhibitor, PF-429242, on Dengue Virus Propagation. Viruses 2016, 8, 46. [Google Scholar] [CrossRef] [Green Version]
- Kleinfelter, L.M.; Jangra, R.K.; Jae, L.T.; Herbert, A.S.; Mittler, E.; Stiles, K.M.; Wirchnianski, A.S.; Kielian, M.; Brummelkamp, T.R.; Dye, J.M.; et al. Haploid Genetic Screen Reveals a Profound and Direct Dependence on Cholesterol for Hantavirus Membrane Fusion. mBio 2015, 6, e00801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasquato, A.; Kunz, S. Novel drug discovery approaches for treating arenavirus infections. Expert Opin. Drug Discov. 2016, 11, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Elagoz, A.; Benjannet, S.; Mammarbassi, A.; Wickham, L.; Seidah, N.G. Biosynthesis and cellular trafficking of the convertase SKI-1/S1P: Ectodomain shedding requires SKI-1 activity. J. Biol. Chem. 2002, 277, 11265–11275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasquato, A.; Cendron, L.; Kunz, S. Cleavage of the Glycoprotein of Arenaviruses. In Activation of Viruses by Host Proteases; Springer: Cham, Switzerland, 2018; pp. 47–70. [Google Scholar]
- Bestle, D.; Heindl, M.R.; Limburg, H.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; Rohde, C.; et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 2020, 3. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.D.; Molloy, S.S.; Jean, F.; Fei, H.; Shimamura, S.; Thomas, G. The ordered and compartment-specfific autoproteolytic removal of the furin intramolecular chaperone is required for enzyme activation. J. Biol. Chem. 2002, 277, 12879–12890. [Google Scholar] [CrossRef] [Green Version]
- Ginefra, P.; Filippi, B.G.H.; Donovan, P.; Bessonnard, S.; Constam, D.B. Compartment-Specific Biosensors Reveal a Complementary Subcellular Distribution of Bioactive Furin and PC7. Cell Rep. 2018, 22, 2176–2189. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Dave, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Yuan, J.; Cai, T.; Zheng, X.; Ren, Y.; Qi, J.; Lu, X.; Chen, H.; Lin, H.; Chen, Z.; Liu, M.; et al. Potentiating CD8(+) T cell antitumor activity by inhibiting PCSK9 to promote LDLR-mediated TCR recycling and signaling. Protein Cell 2021, 12, 240–260. [Google Scholar] [CrossRef]
- He, Z.; Khatib, A.M.; Creemers, J.W.M. Loss of the proprotein convertase Furin in T cells represses mammary tumorigenesis in oncogene-driven triple negative breast cancer. Cancer Lett. 2020, 484, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Rufer, A.C. Drug discovery for enzymes. Drug Discov. Today 2021, 26, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Krammer, F.; Smith, G.J.D.; Fouchier, R.A.M.; Peiris, M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G.; et al. Influenza. Nat. Rev. Dis. Prim. 2018, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Kido, H.; Okumura, Y.; Takahashi, E.; Pan, H.Y.; Wang, S.; Chida, J.; Le, T.Q.; Yano, M. Host envelope glycoprotein processing proteases are indispensable for entry into human cells by seasonal and highly pathogenic avian influenza viruses. J. Mol. Genet. Med. 2008, 3, 167–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| Family | Virus | Capsid | Genome |
|---|---|---|---|
| Retroviridae | HIV, Leukemia viruses | Enveloped | Linear ssRNA(−), RT |
| Flaviridae | HCV, Dengue, Zika, West Nile | Enveloped | Linear ssRNA(+) |
| Togaviridae | Chikungunya | Enveloped | Linear ssRNA(+) |
| Coronaviridae | SARS-CoV-1,2, MERS | Enveloped | Linear ssRNA(+) |
| Filoviridae | Ebola, Marburg | Enveloped | Linear ssRNA(−) |
| Orthomyxoviridae | Avian Influenza H5N1 | Enveloped | Linear ssRNA(−) |
| Paramixoviridae | Measle, RSV, Nipah, MPV | Enveloped | Linear ssRNA(−) |
| Hepadnaviridae | Hepatitis B | Enveloped | Linear ssDNA (−), RT |
| Herpesviridae | Herpes, CMV, Varicella-Zoster | Enveloped | Linear dsDNA |
| Papillomaviridae | HPV | Naked | Circular dsDNA |
| Virus | Glycoprotein | P8 | P6 | P4 | P2 | ↓ | P2′ | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| HIV | gp160 | V | Q | R | E | K | R | A | V | ||
| H7N1 A/FPV/Rostock/34 | HA | K | K | R | E | K | R | G | L | ||
| Avian H5N8 TKY/IRE | HA | R | K | R | K | K | R | G | L | ||
| Avian H5N1 A/HK/97 | HA | R | E | R | R | R | K | K | R | G | L |
| Avian H5N1 TKY/ENG | HA | N | T | P | Q | R | K | K | R | G | L |
| Human CMV | gB | H | N | R | T | K | R | S | T | ||
| Human MPV | F Protein | N | P | R | Q | S | R | F | V | ||
| Human RSV | F Protein | K | K | R | K | R | R | F | L | ||
| Dengue Virus (DENG2) | PrM | H | R | R | E | K | R | S | V | ||
| Ebola Virus | gp160 | G | R | R | T | R | R | E | A | ||
| Chikungunya (CHIKV) | E3E2 | P | R | R | Q | R | R | S | I | ||
| Zika Virus | PrM | A | R | R | S | R | R | A | V | ||
| SARS-CoV-2 | S | S | P | R | R | A | R | S | V | ||
| Variant | First Identification | S-Protein Mutations |
|---|---|---|
| B.1.1.7 α-variant | UK September 2020 | del69-70 HV, del144Y, N501Y, A570D, D614G, P681H, T761I, S982A, D1118H |
| B.1.351 β-variant | South Africa October 2020 | K417N, E484K, N501Y, D614G, A701V |
| B.1.1.248 γ-variant | Brazil, Japan January 2021 | L18F, T20N, P26S, D138Y, R190S, K417T, E484K, N501Y, H655Y, T1027I |
| B.1.167 δ-variant | India December 2020 | T95I, G142D, E154K, K417N, L452R, E484Q, D614G, P681R |
| Substrate | P8 | P6 | P4 | P2 | ↓ | P2′ | P4′ | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CELLULAR | h Pro-SKI-1 site B | R | K | V | F | R | S | L | K | Y | A | E | S |
| h Pro-SKI-1 site B’ | V | T | P | Q | R | K | V | F | R | S | L | K | |
| h Pro-SKI-1 site C | R | H | S | S | R | R | L | L | R | A | I | P | |
| h SREBP2 | S | G | S | G | R | S | V | L | S | F | E | S | |
| h SREBP1 | H | S | P | G | R | N | V | L | G | T | E | S | |
| h ATF6 | A | N | Q | R | R | H | L | L | G | F | S | A | |
| h Luman | G | V | L | S | R | Q | L | R | A | L | P | S | |
| m OASIS (CREB3L1) | Q | M | P | S | R | S | L | L | F | Y | D | D | |
| h CREB-H | R | V | F | S | R | T | L | H | N | D | A | A | |
| h pro-BDNF | K | A | G | S | R | G | L | T | S | L | A | D | |
| h α/β-GlcNAc-1-pTr | K | N | T | G | R | Q | L | K | D | T | F | A | |
| h FAM20C | K | H | T | L | R | I | L | Q | D | F | S | S | |
| h pro-Renin receptor | I | R | K | T | R | T | I | L | E | A | K | Q | |
| VIRAL | Lassa Virus (LASV) GP-C | I | Y | I | S | R | R | L | L | G | T | F | T |
| CCHFV PreGn | S | S | G | S | R | R | L | L | S | E | E | S | |
| LCMV GP-C | K | F | L | T | R | R | L | A | G | T | F | T | |
| Junin Virus (JUNV) GP-C | Q | L | P | R | R | S | L | K | A | F | F | S | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seidah, N.G.; Pasquato, A.; Andréo, U. How Do Enveloped Viruses Exploit the Secretory Proprotein Convertases to Regulate Infectivity and Spread? Viruses 2021, 13, 1229. https://doi.org/10.3390/v13071229
Seidah NG, Pasquato A, Andréo U. How Do Enveloped Viruses Exploit the Secretory Proprotein Convertases to Regulate Infectivity and Spread? Viruses. 2021; 13(7):1229. https://doi.org/10.3390/v13071229
Chicago/Turabian StyleSeidah, Nabil G., Antonella Pasquato, and Ursula Andréo. 2021. "How Do Enveloped Viruses Exploit the Secretory Proprotein Convertases to Regulate Infectivity and Spread?" Viruses 13, no. 7: 1229. https://doi.org/10.3390/v13071229
APA StyleSeidah, N. G., Pasquato, A., & Andréo, U. (2021). How Do Enveloped Viruses Exploit the Secretory Proprotein Convertases to Regulate Infectivity and Spread? Viruses, 13(7), 1229. https://doi.org/10.3390/v13071229