
Coxsackievirus B3 Exploits the Ubiquitin-Proteasome System to Facilitate Viral Replication


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transient Transfection
2.2. Virus Stocks
2.3. Assay of Cell Viability
2.4. Protein Extraction and Anti-HA Pull-Down
2.5. Differential Centrifugation
2.6. Immunoblotting
2.7. Ellman’s Test
2.8. OxyBlot
3. Results
3.1. Evasion of Proteasomal Degradation at CVB3-Utilized Membranes
3.2. Accumulation of Ubiquitin Conjugates as a Result of Membrane Modifications
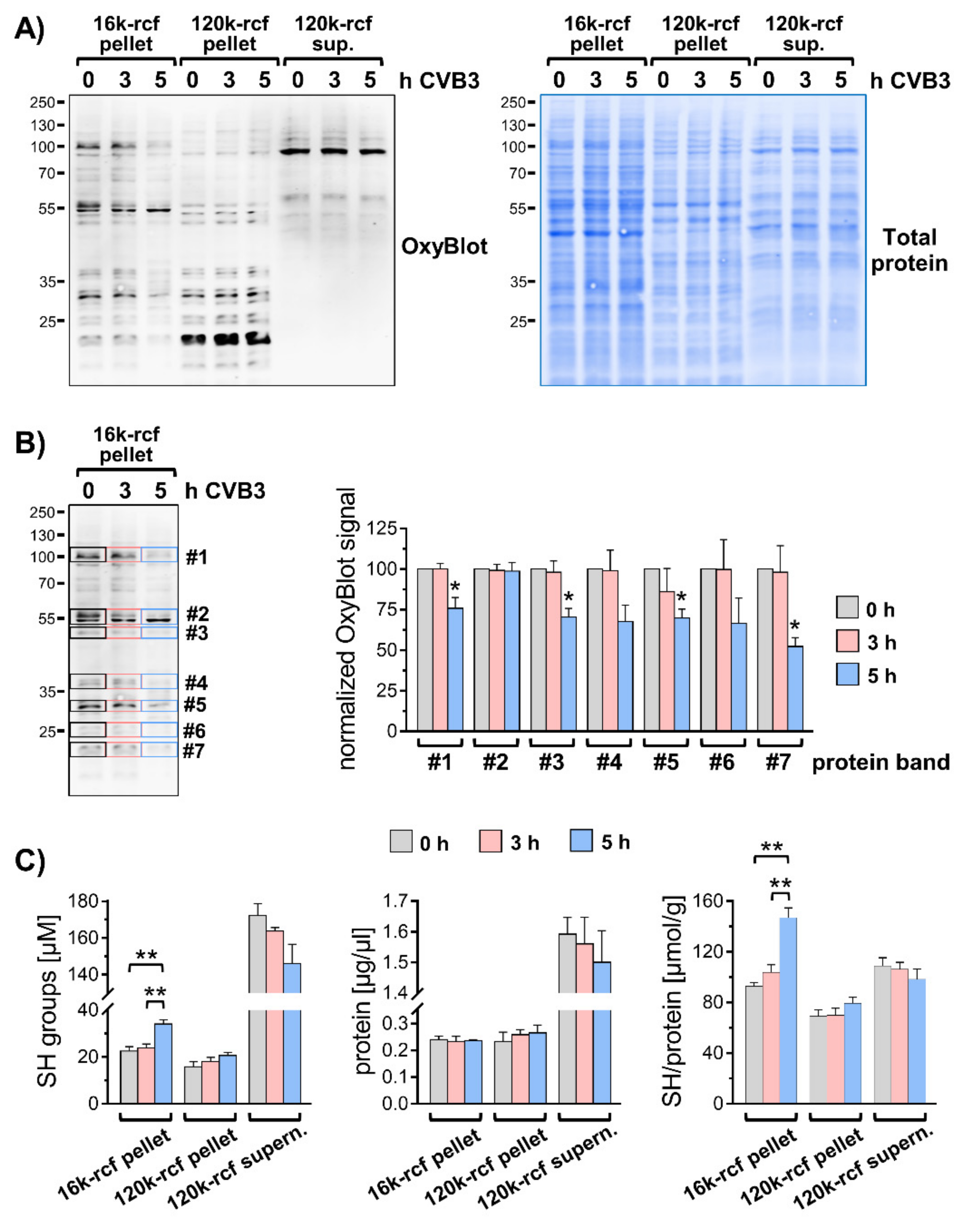
3.3. Modified Redox Homeostasis at Virus-Utilized Membranes
3.4. The Effect of the Proteasome Inhibitor Epoxomicin Is Diminished during CVB3 Infection
3.5. Inhibition of the Proteasome Affects CVB3 Polyprotein Processing
4. Discussion
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Romeo, J.R. Human Enteroviruses in Infectious Diseases; Elsevier/Saunders: Philadelphia, PA, USA, 2017; pp. 1406–1416. [Google Scholar]
- Sin, J.; Mangale, V.; Thienphrapa, W.; Gottlieb, R.A.; Feuer, R. Recent progress in understanding coxsackievirus replication, dissemination, and pathogenesis. Virology 2015, 484, 288–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baggen, J.; Thibaut, H.J.; Strating, J.; Van Kuppeveld, F.J.M. The life cycle of non-polio enteroviruses and how to target it. Nat. Rev. Genet. 2018, 16, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Pohl, C.; Dikic, I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 2019, 366, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Kulathu, Y.; Komander, D. Atypical ubiquitylation—The unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat. Rev. Mol. Cell Biol. 2012, 13, 508–523. [Google Scholar] [CrossRef] [PubMed]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Zhang, J.; Cheung, C.; Suarez, A.; McManus, B.M.; Yang, D. Proteasome Inhibition Reduces Coxsackievirus B3 Replication in Murine Cardiomyocytes. Am. J. Pathol. 2003, 163, 381–385. [Google Scholar] [CrossRef] [Green Version]
- Si, X.; Gao, G.; Wong, J.; Wang, Y.; Zhang, J.; Luo, H. Ubiquitination Is Required for Effective Replication of Coxsackievirus B3. PLoS ONE 2008, 3, e2585. [Google Scholar] [CrossRef]
- Limpens, R.; Van Der Schaar, H.M.; Kumar, D.; Koster, A.; Snijder, E.; Van Kuppeveld, F.J.M.; Bárcena, M. The Transformation of Enterovirus Replication Structures: A Three-Dimensional Study of Single- and Double-Membrane Compartments. mBio 2011, 2, e00166-11. [Google Scholar] [CrossRef] [Green Version]
- Harak, C.; Lohmann, V. Ultrastructure of the replication sites of positive-strand RNA viruses. Virology 2015, 479–480, 418–433. [Google Scholar] [CrossRef] [Green Version]
- Cornell, C.T.; Kiosses, W.B.; Harkins, S.; Whitton, J.L. Inhibition of Protein Trafficking by Coxsackievirus B3: Multiple Viral Proteins Target a Single Organelle. J. Virol. 2006, 80, 6637–6647. [Google Scholar] [CrossRef] [Green Version]
- Van der Schaar, H.M.; Dorobantu, C.M.; Albulescu, L.; Strating, J.; van Kuppeveld, F.J. Fat(al) attraction: Picornaviruses Usurp Lipid Transfer at Membrane Contact Sites to Create Replication Organelles. Trends Microbiol. 2016, 24, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Emmerich, C.H.; Cohen, P. Optimising methods for the preservation, capture and identification of ubiquitin chains and ubiquitylated proteins by immunoblotting. Biochem. Biophys. Res. Commun. 2015, 466, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Hsu, N.-Y.; Ilnytska, O.; Belov, G.; Santiana, M.; Chen, Y.-H.; Takvorian, P.M.; Pau, C.; Van Der Schaar, H.; Kaushik-Basu, N.; Balla, T.; et al. Viral Reorganization of the Secretory Pathway Generates Distinct Organelles for RNA Replication. Cell 2010, 141, 799–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubánková, A.; Humpolickova, J.; Klima, M.; Boura, E. Negative charge and membrane-tethered viral 3B cooperate to recruit viral RNA dependent RNA polymerase 3D pol. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Fujita, K.; Krishnakumar, S.; Franco, D.; Paul, A.V.; London, E.; Wimmer, E. Membrane Topography of the Hydrophobic Anchor Sequence of Poliovirus 3A and 3AB Proteins and the Functional Effect of 3A/3AB Membrane Association upon RNA Replication. Biochemistry 2007, 46, 5185–5199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Zou, Z.; Jiang, Z.; Huang, X.; Liu, Q. Biological Function and Application of Picornaviral 2B Protein: A New Target for Antiviral Drug Development. Viruses 2019, 11, 510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lama, J.; Carrasco, L. Expression of poliovirus nonstructural proteins in Escherichia coli cells. Modification of membrane permeability induced by 2B and 3A. J. Biol. Chem. 1992, 267, 15932–15947. [Google Scholar] [CrossRef]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [Green Version]
- Jung, T.; Höhn, A.; Grune, T. The proteasome and the degradation of oxidized proteins: Part II—Protein oxidation and proteasomal degradation. Redox Biol. 2014, 2, 99–104. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.-C.; Liu, Y.; Wang, C.; Strauss, M.; Rehage, N.; Chen, Y.-H.; Altan-Bonnet, N.; Hogle, J.; Wimmer, E.; Mueller, S.; et al. An Interaction between Glutathione and the Capsid Is Required for the Morphogenesis of C-Cluster Enteroviruses. PLoS Pathog. 2014, 10, e1004052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thibaut, H.J.; Van Der Linden, L.; Jiang, P.; Thys, B.; Canela, M.-D.; Aguado, L.; Rombaut, B.; Wimmer, E.; Paul, A.; Perez-Perez, M.-J.; et al. Binding of Glutathione to Enterovirus Capsids Is Essential for Virion Morphogenesis. PLoS Pathog. 2014, 10, e1004039. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.H.; Sanyal, S. Manipulation of autophagy by (+) RNA viruses. Semin. Cell Dev. Biol. 2020, 101, 3–11. [Google Scholar] [CrossRef]
- Dougherty, S.E.; Maduka, A.O.; Inada, T.; Silva, G.M. Expanding Role of Ubiquitin in Translational Control. Int. J. Mol. Sci. 2020, 21, 1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Wang, Y.; Lin, L.; Si, X.; Wang, T.; Zhong, X.; Tong, L.; Luan, Y.; Chen, Y.; Li, X.; et al. Protease 2A induces stress granule formation during coxsackievirus B3 and enterovirus 71 infections. Virol. J. 2014, 11, 192. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.; Luo, H. The ubiquitin–proteasome pathway in viral infectionsThis paper is one of a selection of papers published in this Special Issue, entitled Young Investigator’s Forum. Can. J. Physiol. Pharmacol. 2006, 84, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zheng, Z.; Shu, B.; Meng, J.; Zhang, Y.; Zheng, C.; Ke, X.; Gong, P.; Hu, Q.; Wang, H. SUMO Modification Stabilizes Enterovirus 71 Polymerase 3D To Facilitate Viral Replication. J. Virol. 2016, 90, 10472–10485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.-C.; Chang, L.-Y.; Wang, Y.-W.; Chen, Y.-C.; Weng, K.-F.; Shih, S.-R.; Shih, H.-M. Sumoylation-promoted Enterovirus 71 3C Degradation Correlates with a Reduction in Viral Replication and Cell Apoptosis. J. Biol. Chem. 2011, 286, 31373–31384. [Google Scholar] [CrossRef] [Green Version]
- Fan, W.; Mar, K.B.; Sari, L.; Gaszek, I.K.; Cheng, Q.; Evers, B.M.; Shelton, J.M.; Wight-Carter, M.; Siegwart, D.J.; Lin, M.M.; et al. TRIM7 inhibits enterovirus replication and promotes emergence of a viral variant with increased pathogenicity. Cell 2021. [Google Scholar] [CrossRef]
- Si, X.; McManus, B.M.; Zhang, J.; Yuan, J.; Cheung, C.; Esfandiarei, M.; Suarez, A.; Morgan, A.; Luo, H. Pyrrolidine Dithiocarbamate Reduces Coxsackievirus B3 Replication through Inhibition of the Ubiquitin-Proteasome Pathway. J. Virol. 2005, 79, 8014–8023. [Google Scholar] [CrossRef] [Green Version]
- Peischard, S.; Ho, H.T.; Theiss, C.; Strutz-Seebohm, N.; Seebohm, G. A Kidnapping Story: How Coxsackievirus B3 and Its Host Cell Interact. Cell. Physiol. Biochem. 2019, 53, 121–140. [Google Scholar] [CrossRef]
- Belov, G.; van Kuppeveld, F.J. (+)RNA viruses rewire cellular pathways to build replication organelles. Curr. Opin. Virol. 2012, 2, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Belov, G.A.; Sztul, E. Rewiring of Cellular Membrane Homeostasis by Picornaviruses. J. Virol. 2014, 88, 9478–9489. [Google Scholar] [CrossRef] [Green Version]
- Greninger, A.L. Picornavirus–Host Interactions to Construct Viral Secretory Membranes. Prog. Mol. Biol. Trans. Sci. 2015, 129, 189–212. [Google Scholar] [CrossRef]
- Zientara-Rytter, K.; Subramani, S. The Roles of Ubiquitin-Binding Protein Shuttles in the Degradative Fate of Ubiquitinated Proteins in the Ubiquitin-Proteasome System and Autophagy. Cells 2019, 8, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vainshtein, A.; Grumati, P. Selective Autophagy by Close Encounters of the Ubiquitin Kind. Cells 2020, 9, 2349. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Wong, J.; Piesik, P.; Fung, G.; Zhang, J.; Jagdeo, J.; Li, X.; Jan, E.; Luo, H. Cleavage of sequestosome 1/p62 by an enteroviral protease results in disrupted selective autophagy and impaired NFKB signaling. Autophagy 2013, 9, 1591–1603. [Google Scholar] [CrossRef] [Green Version]
- Mohamud, Y.; Shi, J.; Qu, J.; Poon, T.; Xue, Y.C.; Deng, H.; Zhang, J.; Luo, H. Enteroviral Infection Inhibits Autophagic Flux via Disruption of the SNARE Complex to Enhance Viral Replication. Cell Rep. 2018, 22, 3292–3303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, A.G.; Wong, J.; Marchant, D.; Luo, H. The ubiquitin-proteasome system in positive-strand RNA virus infection. Rev. Med. Virol. 2012, 23, 85–96. [Google Scholar] [CrossRef]
- Beling, A.; Kespohl, M. Proteasomal Protein Degradation: Adaptation of Cellular Proteolysis with Impact on Virus—and Cytokine-Mediated Damage of Heart Tissue During Myocarditis. Front. Immunol. 2018, 9, 2620. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voss, M.; Braun, V.; Bredow, C.; Kloetzel, P.-M.; Beling, A. Coxsackievirus B3 Exploits the Ubiquitin-Proteasome System to Facilitate Viral Replication. Viruses 2021, 13, 1360. https://doi.org/10.3390/v13071360
Voss M, Braun V, Bredow C, Kloetzel P-M, Beling A. Coxsackievirus B3 Exploits the Ubiquitin-Proteasome System to Facilitate Viral Replication. Viruses. 2021; 13(7):1360. https://doi.org/10.3390/v13071360
Chicago/Turabian StyleVoss, Martin, Vera Braun, Clara Bredow, Peter-Michael Kloetzel, and Antje Beling. 2021. "Coxsackievirus B3 Exploits the Ubiquitin-Proteasome System to Facilitate Viral Replication" Viruses 13, no. 7: 1360. https://doi.org/10.3390/v13071360