
Identification, Genetic Characterization and Validation of Highly Diverse HIV-1 Viruses for Reference Panel Development

 , , , , , , ,
, , , , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Collection and Cell Culture
2.2. Specimens Characterization
2.2.1. Viral Load (VL), p24, and AlphaLISA Assays
2.2.2. RNA Extraction, RT-PCR, and Sequencing
2.2.3. Bioinformatics Data Analysis
3. Results
3.1. Virus Culture and HIV-1 Storage
3.2. HIV-1 Viruses Represent a High Viral Load and p24 Antigen
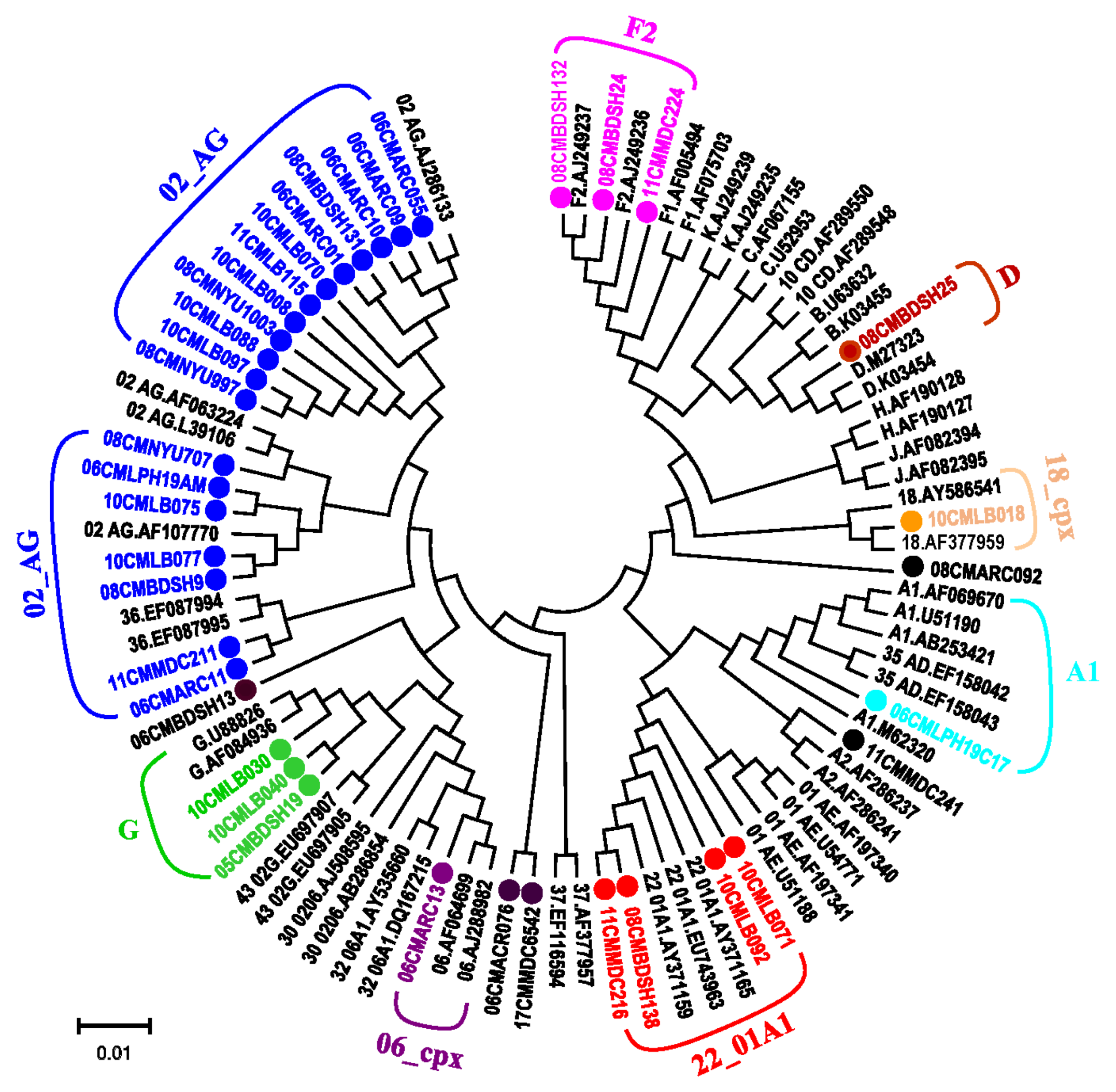
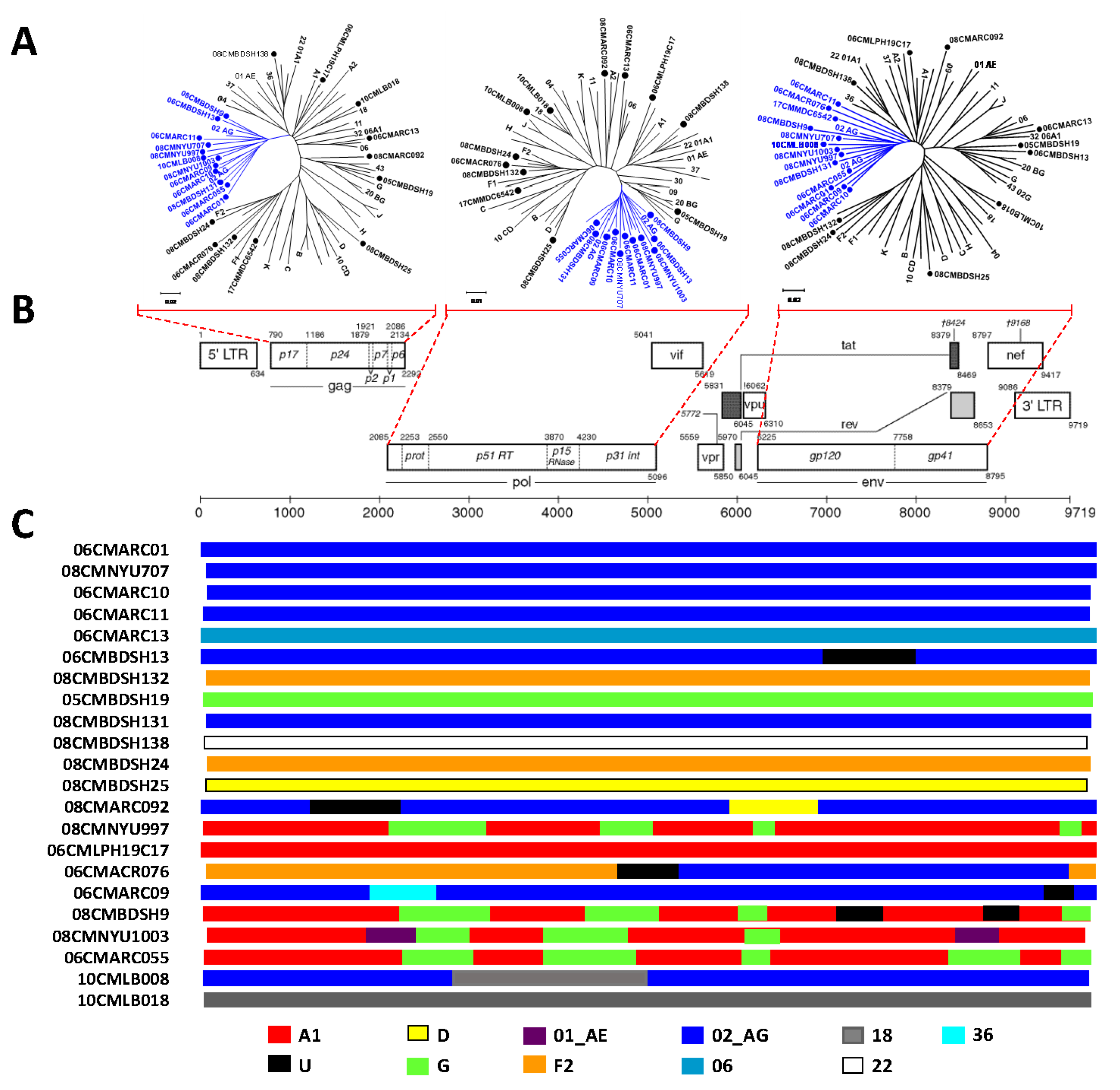
3.3. Phylogenetic Analysis Determines Diverse HIV-1 Viruses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Hora, B.; Keating, S.; Chen, Y.; Sanchez, A.M.; Sabino, E.; Hunt, G.; Ledwaba, J.; Hackett, J.; Swanson, P.; Hewlett, I.; et al. Genetic Characterization of a Panel of Diverse HIV-1 Isolates at Seven International Sites. PLoS ONE 2016, 11, e0157340. [Google Scholar] [CrossRef]
- Sanchez, A.M.; DeMarco, C.T.; Hora, B.; Keinonen, S.; Chen, Y.; Brinkley, C.; Stone, M.; Tobler, L.; Keating, S.; Schito, M.; et al. Development of a contemporary globally diverse HIV viral panel by the EQAPOL program. J. Immunol. Methods 2014, 409, 117–130. [Google Scholar] [CrossRef]
- Manak, M.; Sina, S.; Anekella, B.; Hewlett, I.; Sanders-Buell, E.; Ragupathy, V.; Kim, J.; Vermeulen, M.; Stramer, S.L.; Sabino, E.; et al. Pilot Studies for Development of an HIV Subtype Panel for Surveillance of Global Diversity. Aids Res. Hum. Retrovir. 2012, 28, 594–606. [Google Scholar] [CrossRef]
- Price, M.A.; Rida, W.; Kilembe, W.; Karita, E.; Inambao, M.; Ruzagira, E.; Kamali, A.; Sanders, E.J.; Anzala, O.; Hunter, E.; et al. HIV-1 viral control varies by viral subtype in a large cohort of African adults with incident HIV-1 infection. J. Infect. Dis. 2019, 220, 432–441. [Google Scholar] [CrossRef]
- Banin, A.N.; Tuen, M.; Bimela, J.S.; Tongo, M.; Zappile, P.; Khodadadi-Jamayran, A.; Nanfack, A.J.; Okonko, I.O.; Meli, J.; Wang, X.; et al. Near full genome characterization of HIV-1 unique recombinant forms in Cameroon reveals dominant CRF02_AG and F2 recombination patterns. J. Int. AIDS Soc. 2019, 22, e25362. [Google Scholar] [CrossRef] [PubMed]
- Carr, J.K.; Torimiro, J.N.; Wolfe, N.D.; Eitel, M.N.; Kim, B.; Sanders-Buell, E.; Jagodzinski, L.L.; Gotte, D.; Burke, D.S.; Birx, D.L.; et al. The AG recombinant IbNG and novel strains of group M HIV-1 are common in Cameroon. Virology 2001, 286, 168–181. [Google Scholar] [CrossRef][Green Version]
- Gao, F.; Bailes, E.; Robertson, D.L.; Chen, Y.; Rodenburg, C.M.; Michael, S.F.; Cummins, L.B.; Arthur, L.O.; Peeters, M.; Shaw, G.M.; et al. Origin of HIV-1 in the chimpanzee Pan troglodytes troglodytes. Nat. Cell Biol. 1999, 397, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Hahn, B.H.; Shaw, G.M.; De Cock, K.M.; Sharp, P.M. AIDS as a zoonosis: Scientific and public health implications. Science 2000, 287, 607–614. [Google Scholar] [CrossRef]
- Keele, B.F.; Van Heuverswyn, F.; Li, Y.; Bailes, E.; Takehisa, J.; Santiago, M.L.; Bibollet-Ruche, F.; Chen, Y.; Wain, L.; Liegeois, F.; et al. Chimpanzee Reservoirs of Pandemic and Nonpandemic HIV-1. Science 2006, 313, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Plantier, J.-C.; Leoz, M.; Dickerson, J.E.; De Oliveira, F.; Cordonnier, F.; Lemée, V.; Damond, F.; Robertson, D.L.; Simon, F. A new human immunodeficiency virus derived from gorillas. Nat. Med. 2009, 15, 871–872. [Google Scholar] [CrossRef]
- Powell, R.L.; Zhao, J.; Konings, F.A.; Tang, S.; Ewane, L.; Burda, S.; Urbanski, M.M.; Saa, D.; Hewlett, I.; Nyambi, P.N. Circulating Recombinant Form (CRF) 37_cpx: An Old Strain in Cameroon Composed of Diverse, Genetically Distant Lineages of Subtypes A and G. AIDS Res. Hum. Retrovir. 2007, 23, 923–933. [Google Scholar] [CrossRef]
- Powell, R.L.; Zhao, J.; Konings, F.A.; Tang, S.; Nanfack, A.; Burda, S.; Urbanski, M.M.; Saa, D.; Hewlett, I.; Nyambi, P.N. Identification of a Novel Circulating Recombinant Form (CRF) 36_cpx in Cameroon That Combines Two CRFs (01_AE and 02_AG) with Ancestral Lineages of Subtypes A and G. AIDS Res. Hum. Retrovir. 2007, 23, 1008–1019. [Google Scholar] [CrossRef]
- Vergne, L.; Bourgeois, A.; Mpoudi-Ngole, E.; Mougnutou, R.; Mbuagbaw, J.; Liegeois, F.; Laurent, C.; Butel, C.; Zekeng, L.; Delaporte, E.; et al. Biological and genetic characteristics of HIV infections in Cameroon reveals dual group M and O infections and a correlation between SI-inducing phenotype of the predominant CRF02_AG variant and disease stage. Virology 2003, 310, 254–266. [Google Scholar] [CrossRef]
- Wilbe, K.; Casper, C.; Albert, J.; Leitner, T. Identification of Two CRF11-cpx Genomes and Two Preliminary Representatives of a New Circulating Recombinant Form (CRF13-cpx) of HIV Type 1 in Cameroon. AIDS Res. Hum. Retrovir. 2002, 18, 849–856. [Google Scholar] [CrossRef]
- Zhao, J.; Tang, S.; Ragupathy, V.; Carr, J.K.; Wolfe, N.D.; Awazi, B.; Hewlett, I. Identification and Genetic Characterization of a Novel CRF22_01A1 Recombinant Form of HIV Type 1 in Cameroon. AIDS Res. Hum. Retrovir. 2010, 26, 1033–1045. [Google Scholar] [CrossRef]
- Zhong, P.; Burda, S.; Urbanski, M.; Kenfack, H.; Tongo, M.; Heyndrickx, L.; Nanfack, A.; Shang, J.; Agyingi, L.; Zolla-Pazner, S.; et al. HIV type 1 group M clades infecting subjects from rural villages in equatorial rain forests of Cameroon. J. Acquir. Immune Defic. Syndr. 2002, 31, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, J.; Bodelle, P.; Vallari, A.S.; Coffey, R.; McArthur, C.P.; Schochetman, G.; Devare, S.G.; Brennan, C.A. HIV infections in northwestern Cameroon: Identification of HIV type 1 group O and dual HIV type 1 group M and group O infections. AIDS Res. Hum. Retrovir. 2004, 20, 944–957. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, J.; Badreddine, S.; Swanson, P.; Bodelle, P.; Devare, S.G.; Brennan, C.A. Identification of New CRF43_02G and CRF25_cpx in Saudi Arabia Based on Full Genome Sequence Analysis of Six HIV Type 1 Isolates. AIDS Res. Hum. Retrovir. 2008, 24, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Yebra, G.; Rivas, P.; Herrero, M.D.; López, M.; De Mulder, M.; Puente, S.; Ramírez-Olivencia, G.; Soriano, V.; Holguín, A. Clinical Differences and Viral Diversity between Newly HIV Type 1-Diagnosed African and Non-African Patients in Spain (2005–2007). AIDS Res. Hum. Retrovir. 2009, 25, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Pyne, M.T.; Hackett, J.; Holzmayer, V.; Hillyard, D.R. Large-Scale Analysis of the Prevalence and Geographic Distribution of HIV-1 Non-B Variants in the United States. J. Clin. Microbiol. 2013, 51, 2662–2669. [Google Scholar] [CrossRef] [PubMed]
- Swanson, P.; de Mendoza, C.; Joshi, Y.; Golden, A.; Hodinka, R.L.; Soriano, V.; Devare, S.G.; Hackett, J., Jr. Impact of Human Immunodeficiency Virus Type 1 (HIV-1) Genetic Diversity on Performance of Four Commercial Viral Load Assays: LCx HIV RNA Quantitative, AMPLICOR HIV-1 MONITOR v1.5, VERSANT HIV-1 RNA 3.0, and NucliSens HIV-1 QT. J. Clin. Microbiol 2005, 43, 3860–3868. [Google Scholar] [CrossRef][Green Version]
- Church, D.; Gregson, D.; Lloyd, T.; Klein, M.; Beckthold, B.; Laupland, K.; Gill, M.J. Comparison of the RealTime HIV-1, COBAS TaqMan 48 v1.0, Easy Q v1.2, and Versant v3.0 assays for Determination of HIV-1 Viral Loads in a Cohort of Canadian Patients with Diverse HIV Subtype Infections. J. Clin. Microbiol. 2010, 49, 118–124. [Google Scholar] [CrossRef]
- Gueudin, M.; Plantier, J.-C.; Lemée, V.; Schmitt, M.P.; Chartier, L.; Bourlet, T.; Ruffault, A.; Damond, F.; Vray, M.; Simon, F. Evaluation of the Roche Cobas TaqMan and Abbott Real Time Extraction-Quantification Systems for HIV-1 Subtypes. JAIDS J. Acquir. Immune Defic. Syndr. 2007, 44, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; Sobieszczyk, M.E.; McCutchan, F.E.; Hammer, S.M. The Challenge of HIV-1 Subtype Diversity. N. Engl. J. Med. 2008, 358, 1590–1602. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, M.A.; Brenner, B.G. The Impact of HIV Genetic Polymorphisms and Subtype Differences on the Occurrence of Resistance to Antiretroviral Drugs. Mol. Biol. Int. 2012, 2012, 256982. [Google Scholar] [CrossRef] [PubMed]
- Machuca, A.; Tang, S.; Hu, J.; Lee, S.; Wood, O.; Vockley, C.; Vutukuri, S.G.; Deshmukh, R.; Awazi, B.; Hewlett, I. Increased Genetic Diversity and Intersubtype Recombinants of HIV-1 in Blood Donors From Urban Cameroon. JAIDS J. Acquir. Immune Defic. Syndr. 2007, 45, 361–363. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Tang, S.; Ragupathy, V.; Gaddam, D.; Wang, X.; Zhang, P.; Nyambi, P.N.; Hewlett, I. CRF22_01A1 is Involved in the Emergence of New HIV-1 Recombinants in Cameroon. J. Acquir. Immune Defic. Syndr. 2012, 60, 344–350. [Google Scholar] [CrossRef]
- UNAIDS. Propagation of Primary HIV-1 Isolates. NIH AIDS Research & Reference Reagent Program. Available online: https://www.aidsreagent.org/support_docs/virus.pdf (accessed on 20 November 2020).
- Zhang, M.; Huang, Q.; Huang, Y.; Wood, O.; Yuan, W.; Chancey, C.; Daniel, S.; Rios, M.; Hewlett, I.; Clouse, K.A.; et al. beta-estradiol attenuates the anti-HIV-1 efficacy of Stavudine (D4T) in primary PBL. Retrovirology 2008, 5, 82. [Google Scholar] [CrossRef] [PubMed]
- Ragupathy, V.; Zhao, J.; Wood, O.; Tang, S.; Lee, S.; Nyambi, P.; Hewlett, I. Identification of new, emerging HIV-1 unique recombinant forms and drug resistant viruses circulating in Cameroon. Virol. J. 2011, 8, 185. [Google Scholar] [CrossRef]
- APHL. Best Practice Guidance: Specimen and Specimen-Product Storage and Retention. APHL Issues In Brief. Infect. Dis. 2016, 1, 1–6. [Google Scholar]
- Scott, L.E.; Crump, J.A.; Msuya, E.; Morrissey, A.B.; Venter, W.F.; Stevens, W.S. Abbott RealTime HIV-1 m2000rt viral load testing: Manual extraction versus the automated m2000sp extraction. J. Virol. Methods 2011, 172, 78–80. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pulido-Olmo, H.; Rodríguez-Sánchez, E.; García, J.A.N.; Barderas, M.G.; Alvarez-Llamas, G.; Segura, J.; Fernandez-Alfonso, M.S.; Ruilope, L.M.; Ruiz-Hurtado, G. Rapid, Automated, and Specific Immunoassay to Directly Measure Matrix Metalloproteinase-9–Tissue Inhibitor of Metalloproteinase-1 Interactions in Human Plasma Using AlphaLISA Technology: A New Alternative to Classical ELISA. Front. Immunol. 2017, 8, 853. [Google Scholar] [CrossRef] [PubMed]
- CRFs. HIV Sequence Database. Los Alamos National Laboratory, the Circulating Recombinant Forms (CRFs). Available online: https://www.hiv.lanl.gov/content/sequence/HIV/CRFs/CRFs.html (accessed on 29 April 2021).
- Kuiken, T.; Fouchier, R.; Rimmelzwaan, G.; Osterhaus, A. Emerging viral infections in a rapidly changing world. Curr. Opin. Biotechnol. 2003, 14, 641–646. [Google Scholar] [CrossRef] [PubMed]
- NCBI. National Center for Biotechnology Information (NCBI) Genotyping. Available online: https://www.ncbi.nlm.nih.gov/projects/genotyping/formpage.cgi (accessed on 29 April 2021).
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Dudley, J.; Nei, M.; Kumar, S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) Software Version 4. Mol. Biol. Evol. 2007, 24, 1596–1599. [Google Scholar] [CrossRef]
- Davis, C.; Berry, N.; Heath, A.; Holmes, H. An international collaborative study to establish a replacement World Health Organization International Standard for human immunodeficiency virus 1 RNA nucleic acid assays. Vox Sang. 2008, 95, 218–225. [Google Scholar] [CrossRef]
- Holmes, H.; Davis, C.; Heath, A. Development of the 1st International Reference Panel for HIV-1 RNA genotypes for use in nucleic acid-based techniques. J. Virol. Methods 2008, 154, 86–91. [Google Scholar] [CrossRef]
- WHO. World Health Organization HIV Reference Panels. 2001. Available online: https://www.who.int/bloodproducts/cs/031961.pdf (accessed on 29 April 2021).



{kind=link}
{kind=link}
| Patient ID | Year | VL (107 Copies/mL) | p24 (ng/mL) | AlphaLISA (ng/mL) | Assay’s Agreement | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| W1 | W2 | W3 | W4 | W1 | W2 | W3 | W4 | W1 | W2 | W3 | W4 | |||
| 06CMARC01 | 2006 | 1.4 | 92.4 | 322.7 | - | 0.6 | 49.4 | 112.3 | - | 3.9 | 272.3 | 0.0 ** | - | yes |
| 06CMARC07 | 2006 | 0 | 6.1 | 5.6 | - | 2 | 3.2 | 1.9 | - | 23.5 | 34.7 | 21.8 | - | yes |
| 06CMARC09 | 2006 | 147.2 | 80.8 | 64.8 | - | 56.5 | 91.4 | 27 | - | 290.4 | 330.1 | 60.4 | - | yes * |
| 06CMARC10 | 2006 | 120.8 | 329.6 | 183.4 | - | 58.5 | 110.1 | 93.6 | - | 130.3 | 0.0 ** | 0.0 ** | - | yes |
| 06CMARC11 | 2006 | 12.6 | 27.4 | 14.7 | - | 5.9 | 14.2 | 6.5 | - | 0.9 | 4 | 1.8 | - | yes |
| 06CMARC13 | 2006 | 0.4 | 1.8 | 1.2 | - | 0.2 | 0.9 | 0.8 | - | 0.3 | 1 | 0.7 | - | yes |
| 08CMBDSH24 | 2008 | 17.5 | 121.6 | 22.5 | - | 8.2 | 10.5 | 13.1 | - | 29.7 | 150.8 | 40.8 | - | yes |
| 06CMLPH19C17 | 2006 | 34.6 | 56.7 | 67.7 | - | 13 | 21.4 | 18.5 | - | 101.6 | 59.8 | 136.2 | - | yes * |
| 06CMLPH19AM | 2006 | N.D. | N.D. | N.D. | - | 1.7 | 63.2 | 107 | - | 13.3 | 128.1 | 253.6 | - | yes |
| 06CMARC071 | 2006 | 25 | - | 65.3 | - | 8 | - | 28.2 | - | 58.3 | - | 8.4 | - | yes * |
| 06CMARC076 | 2006 | 25.2 | - | 111.7 | - | 9.5 | - | 41.2 | - | 57.6 | - | 1134 | - | yes |
| 08CMBDSH131 | 2008 | - | 91.7 | 93.6 | - | - | 24.1 | 26.3 | - | - | 32.8 | 28.7 | - | yes * |
| 05CMBDSH19 | 2005 | - | 0.4 | 2.5 | - | - | 0.2 | 0.8 | - | - | 0.5 | 3.1 | - | yes |
| 08CMNYU707 | 2008 | - | 53.6 | 87.3 | - | - | 25.6 | 35.2 | - | - | 274.7 | 479.1 | - | yes |
| 08CMNYU871 | 2008 | - | 100.5 | 52.8 | - | - | 25.2 | 18 | - | - | 53.2 | 36.1 | - | yes |
| 08CMNYU997 | 2008 | - | 1.5 | 1.8 | - | - | 0.3 | 0.5 | - | - | 1.4 | 2 | - | yes |
| 08CMNYU1003 | 2008 | - | 37.3 | 47.9 | - | - | 19.6 | 24.6 | - | - | 0 | 70.4 | - | yes |
| 10CMLB008 | 2010 | - | - | 41.3 | 14.1 | - | - | 21.1 | 9.1 | - | - | 70.6 | 10.9 | yes |
| 10CMLB022 | 2010 | - | - | 100.6 | 0 | - | - | 131.1 | 37.7 | - | - | 275.1 | 40.7 | yes |
| 10CMLB030 | 2010 | - | - | 83.6 | 85.4 | - | - | 18.6 | 23.4 | - | - | 48.7 | 56.4 | yes |
| 10CMLB031 | 2010 | - | - | 180.8 | 52.8 | - | - | 107 | 32.6 | - | - | 217.5 | 60.9 | yes |
| 10CMLB040 | 2010 | - | - | 162.6 | 48.1 | - | - | 120.9 | 6.1 | - | - | 228.5 | 0 | yes |
| 10CMLB070 | 2010 | - | - | 168.4 | 35.1 | - | - | 133.4 | 39.6 | - | - | 323 | 140.4 | yes |
| 10CMLB075 | 2010 | - | - | 106.4 | 0 | - | - | 98.9 | 32.7 | - | - | 261.4 | 108.9 | yes |
| 10CMLB092 | 2010 | - | - | N.D. | N.D. | - | - | 39.8 | 12.5 | - | - | 271.4 | 41.6 | yes |
| 10CMLB097 | 2010 | - | - | N.D. | N.D. | - | - | 4.2 | 1.7 | - | - | 66.1 | 22.9 | yes |
| 10CMLB034 | 2010 | - | - | 73.6 | - | - | - | 27.8 | - | - | - | 246.9 | - | |
| 10CMLB011 | 2010 | - | - | 17.3 | - | - | - | 16.3 | - | - | - | 414.3 | - | |
| 10CMLB012 | 2010 | - | - | 41.5 | - | - | - | 10.5 | - | - | - | 82.5 | - | |
| 10CMLB029 | 2010 | - | - | 298.6 | - | - | - | 173.2 | - | - | - | 318.8 | - | |
| 10CMLB068 | 2010 | - | - | 78 | - | - | - | 92.9 | - | - | - | 189 | - | |
| 10CMNYU488 | 2010 | - | - | 163.7 | - | - | - | 73.2 | - | - | - | 207.5 | - | |
| 08CMARC092 | 2008 | - | - | 111.7 | - | - | - | 27.7 | - | - | - | 288.4 | - | |
| 08CMBDSH132 | 2008 | - | - | 51.4 | - | - | - | 22.8 | - | - | - | 65.6 | - | |
| 08CMBDSH138 | 2008 | - | - | 20.3 | - | - | - | 15.2 | - | - | - | 68 | - | |
| 08CMBDSH139 | 2008 | - | - | 83.6 | - | - | - | 30.6 | - | - | - | 199.6 | - | |
| 08CMBDSH25 | 2008 | - | - | 398.9 | - | - | - | 85.4 | - | - | - | 580.6 | - | |
| 08CMBDSH9 | 2008 | - | - | 44.2 | - | - | - | 23.8 | - | - | - | 50.9 | - | |
| Patient ID | Year | Cultured (Day) | Size (bp) | Identity (%) | Genotype | GB Acc. No |
|---|---|---|---|---|---|---|
| 06CMARC01 | 2006 | 21 | 9083 | 84.2 | 2 | MN153477 |
| 08CMNYU707 | 2008 | 21 | 8855 | 83.2 | 2 | MN153478 |
| 06CMARC10 | 2006 | 21 | 8677 | 82.1 | 2 | MN153479 |
| 06CMARC11 | 2006 | 22 | 8865 | 85.1 | 2 | MN153480 |
| 06CMARC13 | 2006 | 22 | 8857 | 83.6 | 6 | MN153481 |
| 06CMBDSH13 | 2006 | 14 | 8933 | 83.7 | 02,U | MN153482 |
| 08CMBDSH131 | 2008 | 21 | 8862 | 84 | 2 | MN153486 |
| 08CMBDSH132 | 2008 | 21 | 8853 | 86.1 | F2 | MN153483 |
| 08CMBDSH138 | 2008 | 21 | 8868 | 83.7 | 22 | MN153487 |
| 05CMBDSH19 | 2005 | 22 | 9017 | 83.3 | G | MN153484 |
| 08CMBDSH24 | 2008 | 22 | 8855 | 84.7 | F2 | MN153485 |
| 08CMBDSH25 | 2008 | 21 | 8936 | 87.6 | D | MN153488 |
| 08CMARC092 | 2008 | 21 | 8726 | 85.4 | 02,D,U | MN153489 |
| 08CMNYU997 | 2008 | 21 | 8906 | 84.3 | 02,A1 | MN153490 |
| 06CMLPH19C17 | 2006 | 21 | 8915 | 84.3 | A1 | MN153491 |
| 06CMARC076 | 2006 | 20 | 8690 | 84 | 02,F2,U | MN153492 |
| 06CMARC09 | 2006 | 21 | 8777 | 83.6 | 02,36,U | MN153493 |
| 08CMBDSH9 | 2008 | 21 | 8923 | 84 | 2 | MN153494 |
| 08CMNYU1003 | 2008 | 21 | 8916 | 84.1 | 2 | MN153495 |
| 06CMARC055 | 2006 | 8 | 8893 | 84.7 | 02,A1 | MN153496 |
| 10CMLB008 | 2010 | 21 | 8910 | 83.1 | 02,18 | MN153497 |
| 10CMLB018 | 2010 | 21 | 8950 | 85 | 18 | MN153475 |
| 10CMLB030 | 2010 | 21 | 8929 | 83.2 | G | MT349406 |
| 10CMLB040 | 2010 | 21 | 9015 | 83.3 | G,BG | MT349407 |
| 10CMLB088 | 2010 | 21 | 8963 | 84.4 | 2 | MT349408 |
| 10CMLB070 | 2010 | 21 | 9103 | 83.6 | 2 | MT349409 |
| 10CMLB071 | 2010 | 21 | 8822 | 83.5 | 22 | MT349410 |
| 10CMLB075 | 2010 | 21 | 8531 | 84.1 | 2 | MT349411 |
| 10CMLB077 | 2010 | 21 | 8886 | 83.9 | 2 | MT349412 |
| 10CMLB092 | 2010 | 21 | 8247 | 82.6 | 22 | MT349413 |
| 10CMLB097 | 2010 | 21 | 8919 | 84.2 | 2 | MT349414 |
| 11CMLB115 | 2011 | 28 | 8868 | 84.7 | 2 | MT349415 |
| 11CMMDC211 | 2011 | 24 | 8888 | 84.2 | 22 | MT349416 |
| 11CMMDC224 | 2011 | 24 | 8854 | 84.4 | F2 | MT349417 |
| 11CMMDC241 | 2011 | 24 | 8768 | 83.4 | A1,U | MT349418 |
| 06CMLPH19AM | 2006 | 21 | 8913 | 84.9 | 2 | MT349419 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, J.; Huang, H.; Lee, S.; Ragupathy, V.; Biswas, S.; Mbondji-wonje, C.; Wang, X.; Jiang, A.; Hewlett, I. Identification, Genetic Characterization and Validation of Highly Diverse HIV-1 Viruses for Reference Panel Development. Viruses 2021, 13, 1417. https://doi.org/10.3390/v13071417
Zhao J, Huang H, Lee S, Ragupathy V, Biswas S, Mbondji-wonje C, Wang X, Jiang A, Hewlett I. Identification, Genetic Characterization and Validation of Highly Diverse HIV-1 Viruses for Reference Panel Development. Viruses. 2021; 13(7):1417. https://doi.org/10.3390/v13071417
Chicago/Turabian StyleZhao, Jiangqin, Hanxia Huang, Sherwin Lee, Viswanath Ragupathy, Santanu Biswas, Christelle Mbondji-wonje, Xue Wang, Alex Jiang, and Indira Hewlett. 2021. "Identification, Genetic Characterization and Validation of Highly Diverse HIV-1 Viruses for Reference Panel Development" Viruses 13, no. 7: 1417. https://doi.org/10.3390/v13071417
APA StyleZhao, J., Huang, H., Lee, S., Ragupathy, V., Biswas, S., Mbondji-wonje, C., Wang, X., Jiang, A., & Hewlett, I. (2021). Identification, Genetic Characterization and Validation of Highly Diverse HIV-1 Viruses for Reference Panel Development. Viruses, 13(7), 1417. https://doi.org/10.3390/v13071417