
A Fully Protective Congenital CMV Vaccine Requires Neutralizing Antibodies to Viral Pentamer and gB Glycoprotein Complexes but a pp65 T-Cell Response Is Not Necessary

Abstract
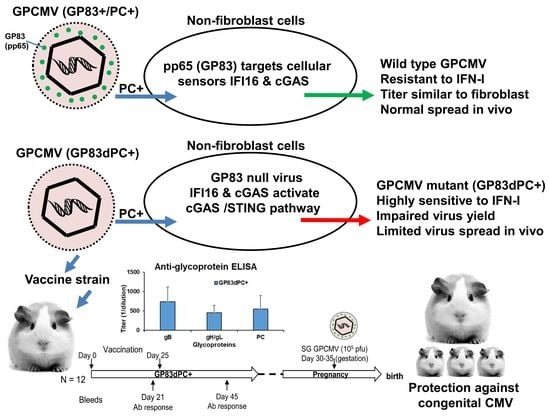
:
1. Introduction
2. Materials and Methods
2.1. Cells, Viruses, and Oligonucleotides
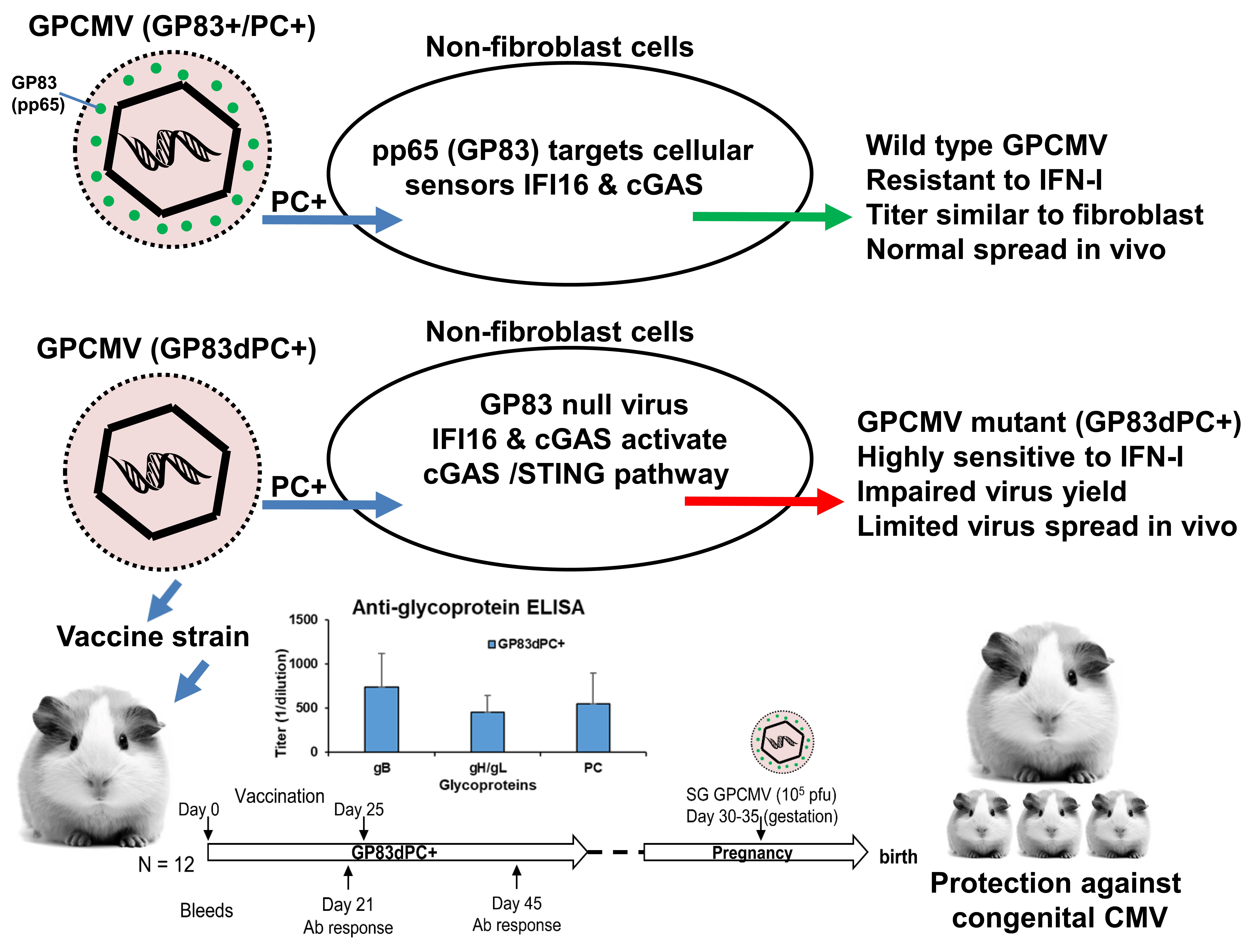
2.2. GP83 Knockout Mutant Construction
2.3. Generation of Gene Mutant GPCMV BACmids, Analysis and Generation of Virus
2.4. Generation of Recombinant Virus
2.5. Ethics
2.6. GPCMV Vaccine Protection Studies
2.6.1. GP83dPC+ Vaccine Protection Study (Pathogenicity)
2.6.2. GP83dPC+ Preconception Vaccine Protection Study
2.6.3. Pathogenicity Study (GP83dPC−)
2.7. Real-Time PCR Assay
2.8. Anti-GPCMV and Individual Glycoprotein Complex Antibody ELISAs
2.8.1. Antibody Avidity Assay
2.8.2. Antibody Depletion from Sera
2.8.3. GPCMV Serum Neutralization
2.9. Statistical Analysis
3. Results
3.1. Generation GP83 Knockout GPCMV Vaccine Strain on Backdrop of PC+/PC− Virus
3.2. Evaluation of Immune Response of GP83dPC+ in Vaccinated Animals
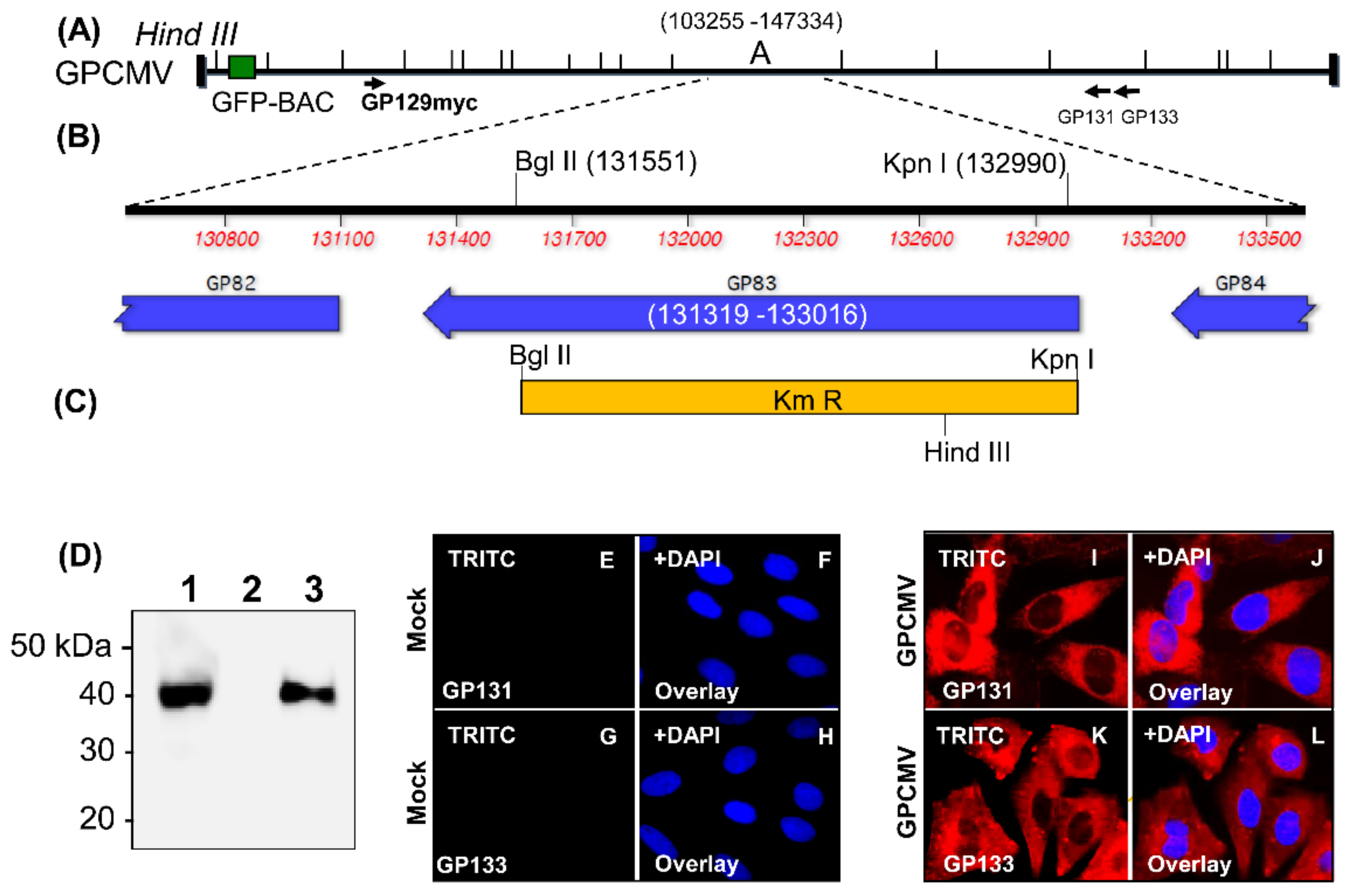
3.3. Comparison of Immune Responses between GP83dPC+ and GP83dPC− Vaccinated Animals
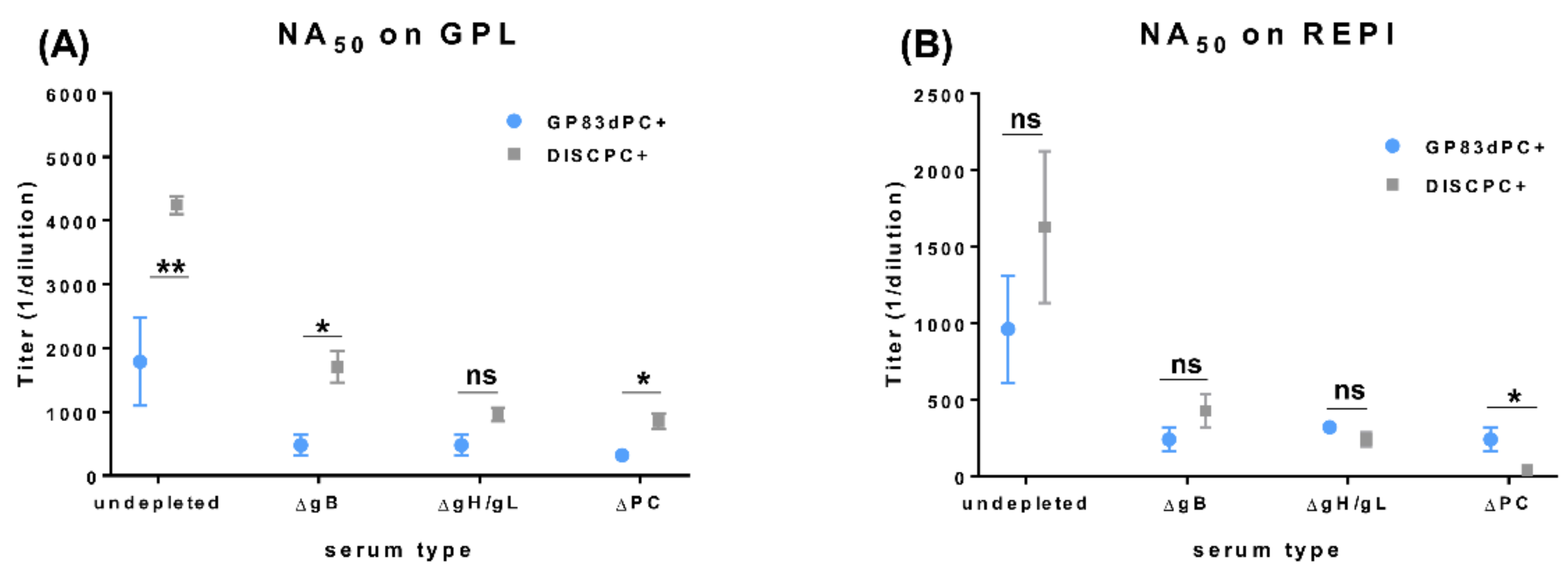
3.4. Depletion of Antibodies to Specific Viral Glycoprotein Complexes Demonstrates Similar Results between GP83dPC+ and DISC (PC+) GPCMV Vaccine Sera
3.5. GP83dPC+ Vaccinated Animals Are Protected against Wild Type Virus Challenge
3.6. Vaccinated Animals Are Protected against Congenital CMV Infection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pass, R.F. Immunization strategy for prevention of congenital cytomegalovirus infection. Infect Agents Dis. 1996, 5, 240–244. [Google Scholar] [PubMed]
- Ross, S.A.; Boppana, S.B. Congenital cytomegalovirus infection: Outcome and diagnosis. Semin. Pediatr. Infect. Dis. 2005, 16, 44–49. [Google Scholar] [CrossRef]
- Dollard, S.C.; Grosse, S.D.; Ross, D.S. New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev. Med. Virol. 2007, 17, 355–363. [Google Scholar] [CrossRef]
- Manicklal, S.; Emery, V.C.; Lazzarotto, T.; Boppana, S.B.; Gupta, R.K. The “silent” global burden of congenital cytomegalovirus. Clin. Microbiol. Rev. 2013, 26, 86–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannon, M.J.; Griffiths, P.D.; Aston, V.; Rawlinson, W.D. Universal newborn screening for congenital CMV infection: What is the evidence of potential benefit? Rev. Med. Virol. 2014, 24, 291–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morton, C.C.; Nance, W.E. Newborn hearing screening—A silent revolution. N. Engl. J. Med. 2006, 354, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Stratton, K.R.; Durch, J.S.; Lawrence, R.S. Vaccines for the 21st Century: A Tool for Decisionmaking; The National Academies Collection Report; Stratton, K.R., Durch, J.S., Lawrence, R.S., Eds.; National Academies Press: Washington, DC, USA, 2000. [Google Scholar]
- Britt, W.J. Congenital Human Cytomegalovirus Infection and the Enigma of Maternal Immunity. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, K.P. Mouse Cytomegalovirus: Placental Infection. J. Infect. Dis. 1969, 120, 445–450. [Google Scholar] [CrossRef]
- Griffith, B.P.; McCormick, S.R.; Fong, C.K.; Lavallee, J.T.; Lucia, H.L.; Goff, E. The placenta as a site of cytomegalovirus infection in guinea pigs. J. Virol. 1985, 55, 402–409. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.L.; Nankervis, G.A. Experimental Congenital Infection with Cytomegalovirus: A Guinea Pig Model. J. Infect. Dis. 1978, 138, 650–654. [Google Scholar] [CrossRef] [PubMed]
- Woolf, N. Guinea pig model of congenital CMV-induced hearing loss: A review. Transplant. Proc. 1991, 23, 32. [Google Scholar] [PubMed]
- McGregor, A.; Choi, K.Y. Cytomegalovirus antivirals and development of improved animal models. Expert Opin. Drug Metab. Toxicol. 2011, 7, 1245–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kern, E.R. Pivotal role of animal models in the development of new therapies for cytomegalovirus infections. Antivir. Res. 2006, 71, 164–171. [Google Scholar] [CrossRef]
- McGregor, A.; McVoy, M.A.; Schleiss, M.R. The Guinea Pig Model of Congenital Cytomegalovirus Infection. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention, 3rd ed.; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; pp. 88–118. [Google Scholar]
- Britt, W.J.; Mach, M. Human Cytomegalovirus Glycoproteins. Intervirology 1996, 39, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Gretch, D.R.; Kari, B.; Gehrz, R.C.; Stinski, M.F. A multigene family encodes the human cytomegalovirus glycoprotein complex gcII (gp 47–52 complex). J. Virol. 1988, 62, 1956–1962. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, L.; Gretch, D.R.; Kari, B.; Gehrz, R.C.; Stinski, M.F. Identification and characterization of three distinct families of glycoprotein complexes in the envelopes of human cytomegalovirus. J. Virol. 1988, 62, 875–881. [Google Scholar]
- Huber, M.T.; Compton, T. The human cytomegalovirus UL74 gene encodes the third component of the glycoprotein H-glycoprotein L-containing envelope complex. J. Virol. 1998, 72, 8191–8197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pass, R.F.; Zhang, C.; Evans, A.; Simpson, T.; Andrews, W.; Huang, M.L.; Corey, L.; Hill, J.; Davis, E.; Flanigan, B.S. Vaccine prevention of maternal cytomegalovirus infection. N. Engl. J. Med. 2009, 360, 1191–1199. [Google Scholar] [CrossRef]
- Shimamura, M.; Mach, M.; Britt, W.J. Human cytomegalovirus infection elicits a glycoprotein M (gM)/gN-specific virus-neutralizing antibody response. J. Virol. 2006, 80, 4591–4600. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.; Wang, S.; Britt, W.J.; Lu, S. DNA vaccines expressing glycoprotein complex II antigens gM and gN elicited neutralizing antibodies against multiple human cytomegalovirus (HCMV) isolates. Vaccine 2007, 25, 3319–3327. [Google Scholar] [CrossRef]
- Nguyen, C.C.; Kamil, J.P. Pathogen at the Gates: Human Cytomegalovirus Entry and Cell Tropism. Viruses 2018, 10, 704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, S.; Hornig, J.; Maddux, S.; Choi, K.Y.; McGregor, A. Viral Glycoprotein Complex Formation, Essential Function and Immunogenicity in the Guinea Pig Model for Cytomegalovirus. PLoS ONE 2015, 10, e0135567. [Google Scholar]
- Coleman, S.; Choi, K.Y.; Root, M.; McGregor, A. A Homolog Pentameric Complex Dictates Viral Epithelial Tropism, Pathogenicity and Congenital Infection Rate in Guinea Pig Cytomegalovirus. PLoS Pathog. 2016, 12, e1005755. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.Y.; Root, M.; McGregor, A. A Novel Non-Replication-Competent Cytomegalovirus Capsid Mutant Vaccine Strategy Is Effective in Reducing Congenital Infection. J. Virol. 2016, 90, 7902–7919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, K.Y.; El-Hamdi, N.S.; McGregor, A. Requirements for guinea pig cytomegalovirus tropism and antibody neutralization on placental amniotic sac cells. J. Gen. Virol. 2020, 101, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.Y.; El-Hamdi, N.S.; McGregor, A. Neutralizing antibodies to gB based CMV vaccine requires full length antigen but reduced virus neutralization on non-fibroblast cells limits vaccine efficacy in the guinea pig model. Vaccine 2020, 38, 2340–2349. [Google Scholar] [CrossRef]
- Rasmussen, L.; Geissler, A.; Cowan, C.; Chase, A.; Winters, M. The Genes Encoding the gCIII Complex of Human Cytomegalovirus Exist in Highly Diverse Combinations in Clinical Isolates. J. Virol. 2002, 76, 10841–10848. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.Y.; El-Hamdi, N.S.; McGregor, A. Convalescent Immunity to Guinea Pig Cytomegalovirus Induces Limited Cross Strain Protection against Re-Infection but High-Level Protection against Congenital Disease. Int. J. Mol. Sci. 2020, 21, 5997. [Google Scholar] [CrossRef]
- Revello, M.G.; Gerna, G. Human cytomegalovirus tropism for endothelial/epithelial cells: Scientific background and clinical implications. Rev. Med. Virol. 2010, 20, 136–155. [Google Scholar] [CrossRef]
- Yamada, S.; Fukuchi, S.; Hashimoto, K.; Fukui, Y.; Tsuda, M.; Kataoka, M.; Katano, H.; Inoue, N. Guinea pig cytomegalovirus GP129/131/133, homologues of human cytomegalovirus UL128/130/131A, are necessary for infection of monocytes and macrophages. J. Gen. Virol. 2014, 95, 1376–1382. [Google Scholar] [CrossRef]
- Coleman, S.; Choi, K.Y.; McGregor, A. Cytomegalovirus UL128 homolog mutants that form a pentameric complex produce virus with impaired epithelial and trophoblast cell tropism and altered pathogenicity in the guinea pig. Virology 2017, 509, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Makino, R.; Yamada, K.; Matsuura, M.; Okumura, M.; Yamada, S.; Watanabe, S.; Inoue, N. Differences in the effects of mutations in GP131, a guinea pig cytomegalovirus homologue of pentameric complex component UL130, on macrophage and epithelial cell infection. J. Gen. Virol. 2018, 99, 1425–1431. [Google Scholar] [CrossRef] [PubMed]
- Kabanova, A.; Marcandalli, J.; Zhou, T.; Bianchi, S.; Baxa, U.; Tsybovsky, Y.; Lilleri, D.; Silacci-Fregni, C.; Foglierini, M.; Fernandez-Rodriguez, B.M.; et al. Platelet-derived growth factor-α receptor is the cellular receptor for human cytomegalovirus gHgLgO trimer. Nat. Microbiol. 2016, 1, 1–8. [Google Scholar] [CrossRef]
- Choi, K.Y.; El-Hamdi, N.S.; McGregor, A. Inclusion of the Viral Pentamer Complex in a Vaccine Design Greatly Improves Protection against Congenital Cytomegalovirus in the Guinea Pig Model. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- El-Hamdi, N.S.; Choi, K.Y.; McGregor, A. Guinea pig cytomegalovirus trimer complex gH/gL/gO uses PDGFRA as universal receptor for cell fusion and entry. Virology 2020, 548, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Prager, A.; Boos, S.; Resch, M.; Brizić, I.; Mach, M.; Wildner, S.; Scrivano, L.; Adler, B. Human cytomegalovirus glycoprotein complex gH/gL/gO uses PDGFR-α as a key for entry. PLoS Pathog. 2017, 13, e1006281. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Martin, N.; Marcandalli, J.; Huang, C.S.; Arthur, C.P.; Perotti, M.; Foglierini, M.; Ho, H.; Dosey, A.M.; Shriver, S.; Payandeh, J.; et al. An Unbiased Screen for Human Cytomegalovirus Identifies Neuropilin-2 as a Central Viral Receptor. Cell 2018, 174, 1158–1171.e19. [Google Scholar] [CrossRef] [Green Version]
- Mori, Y.; Xiaofei, E.; Meraner, P.; Ping, L.; Perreira, J.M.; Aker, A.M.; McDougall, W.M.; Zhuge, R.; Chan, G.C.; Gerstein, R.M.; et al. Faculty Opinions recommendation of OR14I1 is a receptor for the human cytomegalovirus pentameric complex and defines viral epithelial cell tropism. Fac. Opin. Post Publ. Peer Rev. Biomed. Lit. 2019, 116, 7043–7052. [Google Scholar] [CrossRef]
- Vanarsdall, A.L.; Pritchard, S.R.; Wisner, T.W.; Liu, J.; Jardetzky, T.S.; Johnson, D.C. CD147 Promotes Entry of Pentamer-Expressing Human Cytomegalovirus into Epithelial and Endothelial Cells. mBio 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Stein, K.R.; Gardner, T.; Hernandez, R.E.; Kraus, T.A.; Duty, J.A.; Ubarretxena-Belandia, I.; Moran, T.M.; Tortorella, D. CD46 facilitates entry and dissemination of human cytomegalovirus. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Feire, A.L.; Koss, H.; Compton, T. Cellular integrins function as entry receptors for human cytomegalovirus via a highly conserved disintegrin-like domain. Proc. Natl. Acad. Sci. USA 2004, 101, 15470–15475. [Google Scholar] [CrossRef] [Green Version]
- Adler, S.P.; Nigro, G.; Pereira, L. Recent Advances in the Prevention and Treatment of Congenital Cytomegalovirus Infections. Semin. Perinatol. 2007, 31, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Marshall, G.S.; Rabalais, G.P.; Stout, G.G.; Waldeyer, S.L. Antibodies to Recombinant-Derived Glycoprotein B after Natural Human Cytomegalovirus Infection Correlate with Neutralizing Activity. J. Infect. Dis. 1992, 165, 381–384. [Google Scholar] [CrossRef]
- Fowler, K.B.; Stagno, S.; Pass, R.; Britt, W.J.; Boll, T.J.; Alford, C.A. The Outcome of Congenital Cytomegalovirus Infection in Relation to Maternal Antibody Status. N. Engl. J. Med. 1992, 326, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Schleiss, M.R.; Bourne, N.; Stroup, G.; Bravo, F.J.; Jensen, N.J.; Bernstein, D.I. Protection against Congenital Cytomegalovirus Infection and Disease in Guinea Pigs, Conferred by a Purified Recombinant Glycoprotein B Vaccine. J. Infect. Dis. 2004, 189, 1374–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, D.I.; Munoz, F.M.; Callahan, S.T.; Rupp, R.; Wootton, S.H.; Edwards, K.M.; Turley, C.B.; Stanberry, L.R.; Patel, S.M.; Mcneal, M.M.; et al. Safety and efficacy of a cytomegalovirus glycoprotein B (gB) vaccine in adolescent girls: A randomized clinical trial. Vaccine 2016, 34, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, P.D.; Stanton, A.; McCarrell, E.; Smith, C.; Osman, M.; Harber, M.; Davenport, A.; Jones, G.; Wheeler, D.C.; O’Beirne, J.; et al. Cytomegalovirus glycoprotein-B vaccine with MF59 adjuvant in transplant recipients: A phase 2 randomised placebo-controlled trial. Lancet 2011, 377, 1256–1263. [Google Scholar] [CrossRef] [Green Version]
- Harrison, C.J.; Britt, W.J.; Chapman, N.M.; Mullican, J.; Tracy, S. Reduced congenital cytomegalovirus (CMV) infection after maternal immunization with a guinea pig CMV glycoprotein before gestational primary CMV infection in the guinea pig model. J. Infect. Dis. 1995, 172, 1212–1220. [Google Scholar] [CrossRef]
- Schleiss, M.R. Animal models of congenital cytomegalovirus infection: An overview of progress in the characterization of guinea pig cytomegalovirus (GPCMV). J. Clin. Virol. 2002, 25, 37–49. [Google Scholar] [CrossRef]
- Cui, X.; Cao, Z.; Wang, S.; Lee, R.B.; Wang, X.; Murata, H.; Adler, S.P.; McVoy, M.A.; Snapper, C.M. Novel trimeric human cytomegalovirus glycoprotein B elicits a high-titer neutralizing antibody response. Vaccine 2018, 36, 5580–5590. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.Y.; El-Hamdi, N.S.; McGregor, A. A trimeric capable gB CMV vaccine provides limited protection against a highly cell associated and epithelial tropic strain of cytomegalovirus in guinea pigs. J. Gen. Virol. 2021, 102, 1579. [Google Scholar] [CrossRef]
- Cui, X.; Meza, B.P.; Adler, S.P.; McVoy, M.A. Cytomegalovirus vaccines fail to induce epithelial entry neutralizing antibodies comparable to natural infection. Vaccine 2008, 26, 5760–5766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouts, A.E.; Chan, P.; Stephan, J.P.; Vandlen, R.; Feierbach, B. Antibodies against the gH/gL/UL128/UL130/UL131 complex comprise the majority of the anti-cytomegalovirus (anti-CMV) neutralizing antibody response in CMV hyperimmune globulin. J. Virol. 2012, 86, 7444–7447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerna, G.; Lilleri, D.; Rognoni, V.; Agozzino, M.; Meloni, F.; Oggionni, T.; Pellegrini, C.; Arbustini, E.; D’Armini, A.M. Preemptive Therapy for Systemic and Pulmonary Human Cytomegalovirus Infection in Lung Transplant Recipients. Arab. Archaeol. Epigr. 2009, 9, 1142–1150. [Google Scholar] [CrossRef] [PubMed]
- Sylwester, A.W.; Mitchell, B.L.; Edgar, J.B.; Taormina, C.; Pelte, C.; Ruchti, F.; Sleath, P.R.; Grabstein, K.H.; Hosken, N.A.; Kern, F.; et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 2005, 202, 673–685. [Google Scholar] [CrossRef] [Green Version]
- Wills, M.R.; Mason, G.M.; Sissons, J.G.P. Adaptive Cellular Immunity to Human Cytomegalovirus. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; pp. 142–172. [Google Scholar]
- McGregor, A.; Liu, F.; Schleiss, M.R. Molecular, Biological, and In Vivo Characterization of the Guinea Pig Cytomegalovirus (CMV) Homologs of the Human CMV Matrix Proteins pp71 (UL82) and pp65 (UL83). J. Virol. 2004, 78, 9872–9889. [Google Scholar] [CrossRef] [Green Version]
- Hornig, J.; Choi, K.Y.; McGregor, A. The essential role of guinea pig cytomegalovirus (GPCMV) IE1 and IE2 homologs in viral replication and IE1-mediated ND10 targeting. Virology 2017, 504, 122–140. [Google Scholar] [CrossRef]
- Harari, A.; Zimmerli, S.C.; Pantaleo, G. Cytomegalovirus (CMV)-Specific cellular immune responses. Hum. Immunol. 2004, 65, 500–506. [Google Scholar] [CrossRef]
- Chiu, Y.-L.; Lin, C.-H.; Sung, B.-Y.; Chuang, Y.-F.; Schneck, J.P.; Kern, F.; Pawelec, G.; Wang, G.C. Cytotoxic polyfunctionality maturation of cytomegalovirus-pp65-specific CD4 + and CD8 + T-cell responses in older adults positively correlates with response size. Sci. Rep. 2016, 6, 19227. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, A.-H.; Kuo, C.-F.; Chou, I.-J.; Tseng, W.-Y.; Chen, Y.-F.; Yu, K.-H.; Luo, S.-F. Human cytomegalovirus pp65 peptide-induced autoantibodies cross-reacts with TAF9 protein and induces lupus-like autoimmunity in BALB/c mice. Sci. Rep. 2020, 10, 9662. [Google Scholar] [CrossRef]
- Schmolke, S.; Kern, H.F.; Drescher, P.; Jahn, G.; Plachter, B. The dominant phosphoprotein pp65 (UL83) of human cytomegalovirus is dispensable for growth in cell culture. J. Virol. 1995, 69, 5959–5968. [Google Scholar] [CrossRef] [Green Version]
- Biolatti, M.; Dell’Oste, V.; Pautasso, S.; Gugliesi, F.; von Einem, J.; Krapp, C.; Jakobsen, M.R.; Borgogna, C.; Gariglio, M.; De Andrea, M.; et al. Human Cytomegalovirus Tegument Protein pp65 (pUL83) Dampens Type I Interferon Production by Inactivating the DNA Sensor cGAS without Affecting STING. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Biolatti, M.; Dell’Oste, V.; Pautasso, S.; von Einem, J.; Marschall, M.; Plachter, B.; Gariglio, M.; De Andrea, M.; Landolfo, S. Regulatory Interaction between the Cellular Restriction Factor IFI16 and Viral pp65 (pUL83) Modulates Viral Gene Expression and IFI16 Protein Stability. J. Virol. 2016, 90, 8238–8250. [Google Scholar] [CrossRef] [Green Version]
- Schleiss, M.R.; Lacayo, J.C.; Belkaid, Y.; McGregor, A.; Stroup, G.; Rayner, J.; Alterson, K.; Chulay, J.D.; Smith, J.F. Preconceptual Administration of an Alphavirus Replicon UL83 (pp65 Homolog) Vaccine Induces Humoral and Cellular Immunity and Improves Pregnancy Outcome in the Guinea Pig Model of Congenital Cytomegalovirus Infection. J. Infect. Dis. 2007, 195, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.Y.; El-Hamdi, N.; Hornig, J.; McGregor, A. Guinea Pig Cytomegalovirus Protective T Cell Antigen GP83 Is a Functional pp65 Homolog for Innate Immune Evasion and Pentamer-Dependent Virus Tropism. J. Virol. 2021, 95. [Google Scholar] [CrossRef] [PubMed]
- Cardin, R.D.; Bravo, F.J.; Pullum, D.A.; Orlinger, K.; Watson, E.M.; Aspoeck, A.; Fuhrmann, G.; Guirakhoo, F.; Monath, T.; Bernstein, D.I. Replication-defective lymphocytic choriomeningitis virus vectors expressing guinea pig cytomegalovirus gB and pp65 homologs are protective against congenital guinea pig cytomegalovirus infection. Vaccine 2016, 34, 1993–1999. [Google Scholar] [CrossRef] [PubMed]
- Bialas, K.M.; Tanaka, T.; Tran, D.; Varner, V.; De La Rosa, E.C.; Chiuppesi, F.; Wussow, F.; Kattenhorn, L.; Macri, S.; Kunz, E.L.; et al. Maternal CD4+T cells protect against severe congenital cytomegalovirus disease in a novel nonhuman primate model of placental cytomegalovirus transmission. Proc. Natl. Acad. Sci. USA 2015, 112, 13645–13650. [Google Scholar] [CrossRef] [Green Version]
- Abel, K.; Martinez, J.; Yue, Y.; Lacey, S.F.; Wang, Z.; Strelow, L.; Dasgupta, A.; Li, Z.; Schmidt, K.A.; Oxford, K.L.; et al. Vaccine-Induced Control of Viral Shedding following Rhesus Cytomegalovirus Challenge in Rhesus Macaques. J. Virol. 2010, 85, 2878–2890. [Google Scholar] [CrossRef] [Green Version]
- Plotkin, S.A.; Farquhar, J.D.; Ogra, P.L. Immunologic properties of RA27-3 rubella virus vaccine. A comparison with strains presently licensed in the United States. JAMA 1973, 225, 585–590. [Google Scholar] [CrossRef]
- Takahashi, M.; Otsuka, T.; Okuno, Y.; Asano, Y.; Yazaki, T.; Isomura, S. Live Vaccine Used to Prevent the Spread of Varicella in Children in Hospital. Lancet 1974, 304, 1288–1290. [Google Scholar] [CrossRef]
- Schleiss, M.R.; Buus, R.J.; Choi, K.Y.; McGregor, A. An attenuated CMV vaccine with a deletion in tegument protein GP83 (pp65 homolog) protects against placental infection and improves pregnancy outcome in a Guinea pig challenge model. Future Virol. 2013, 8, 1151–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, X.; McGregor, A.; Schleiss, M.R.; McVoy, M.A. Cloning the complete guinea pig cytomegalovirus genome as an infectious bacterial artificial chromosome with excisable origin of replication. J. Virol. Methods 2008, 149, 231–239. [Google Scholar] [CrossRef] [Green Version]
- McGregor, A.; Schleiss, M.R. Molecular Cloning of the Guinea Pig Cytomegalovirus (GPCMV) Genome as an Infectious Bacterial Artificial Chromosome (BAC) in Escherichia coli. Mol. Genet. Metab. 2001, 72, 15–26. [Google Scholar] [CrossRef] [PubMed]
- McGregor, A.; Choi, K.Y.; Cui, X.; McVoy, M.A.; Schleiss, M.R. Expression of the human cytomegalovirus UL97 gene in a chimeric guinea pig cytomegalovirus (GPCMV) results in viable virus with increased susceptibility to ganciclovir and maribavir. Antivir. Res. 2008, 78, 250–259. [Google Scholar] [CrossRef] [Green Version]
- McGregor, A.; Choi, K.; Schleiss, M. Guinea pig cytomegalovirus GP84 is a functional homolog of the human cytomegalovirus (HCMV) UL84 gene that can complement for the loss of UL84 in a chimeric HCMV. J. Virol. 2011, 410, 76–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanarsdall, A.L.; Howard, P.W.; Wisner, T.W.; Johnson, D.C. Human Cytomegalovirus gH/gL Forms a Stable Complex with the Fusion Protein gB in Virions. PLoS Pathog. 2016, 12, e1005564. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Freed, D.C.; He, X.; Li, F.; Tang, A.; Cox, K.S.; Dubey, S.A.; Cole, S.; Medi, M.B.; Liu, Y.; et al. A replication-defective human cytomegalovirus vaccine for prevention of congenital infection. Sci. Transl. Med. 2016, 8, 362ra145. [Google Scholar] [CrossRef]
- Yue, Y.; Wang, Z.; Abel, K.; Li, J.; Strelow, L.; Mandarino, A.; Eberhardt, M.K.; Schmidt, K.A.; Diamond, D.J.; Barry, P.A. Evaluation of recombinant modified vaccinia Ankara virus-based rhesus cytomegalovirus vaccines in rhesus macaques. Med. Microbiol. Immunol. 2008, 197, 117–123. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Snapper, C.M. Development of novel vaccines against human cytomegalovirus. Hum. Vaccines Immunother. 2019, 15, 2673–2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, L.; Piccinini, G.; Lilleri, D.; Revello, M.G.; Wang, Z.; Markel, S.; Diamond, D.; Luzuriaga, K. Human Cytomegalovirus Proteins pp65 and Immediate Early Protein 1 Are Common Targets for CD8+T Cell Responses in Children with Congenital or Postnatal Human Cytomegalovirus Infection. J. Immunol. 2004, 172, 2256–2264. [Google Scholar] [CrossRef] [Green Version]
- Case, R.; Sharp, E.; Benned-Jensen, T.; Rosenkilde, M.M.; Davis-Poynter, N.; Farrell, H.E. Functional analysis of the murine cytomegalovirus chemokine receptor homologue M33: Ablation of constitutive signaling is associated with an attenuated phenotype in vivo. J. Virol. 2008, 82, 1884–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caposio, P.; Worm, S.V.D.; Crawford, L.; Perez, W.; Kreklywich, C.; Gilbride, R.M.; Hughes, C.M.; Ventura, A.B.; Ratts, R.; Marshall, E.E.; et al. Characterization of a live-attenuated HCMV-based vaccine platform. Sci. Rep. 2019, 9, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment Group (n) | Litter Outcomes | Outcomes for Total Pups a (no. [%]) | ||||||
|---|---|---|---|---|---|---|---|---|
| Total Litters | Live Only | Dead Only | Mixed | Pre-Term | Live-Born | Still-Born | Pre-Term b | |
| GP83dPC+ (12) | 12 | 9 | 0 | 2 | 1 | 34 [82.9] | 3 [7.3] | 4 [9.8] |
| No vaccine (14) | 14 | 7 | 4 | 3 | 0 | 27 [56.3] | 21 [43.8] | 0 [0] |
| Vaccine Groups (n) | Number [%] of Pups Positive for GPCMV in Target Organs | Number [%] of CMV + Pups a | |||
|---|---|---|---|---|---|
| Lung | Liver | Spleen | Brain | ||
| GP83dPC+ (41) | 0 | 0 | 0 | 0 | 0 [0] |
| [viral load] | [NVL] b | [NVL] | [NVL] | [NVL] | |
| No vaccine (35) | 21 [60.0] | 15 [42.9] | 19 [54.3] | 18 [51.4] | 28 [80.0] |
| [viral load] c | [4.62 × 102] | [4.25 × 102] | [1.33 × 103] | [1.87 × 103] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, K.Y.; McGregor, A. A Fully Protective Congenital CMV Vaccine Requires Neutralizing Antibodies to Viral Pentamer and gB Glycoprotein Complexes but a pp65 T-Cell Response Is Not Necessary. Viruses 2021, 13, 1467. https://doi.org/10.3390/v13081467
Choi KY, McGregor A. A Fully Protective Congenital CMV Vaccine Requires Neutralizing Antibodies to Viral Pentamer and gB Glycoprotein Complexes but a pp65 T-Cell Response Is Not Necessary. Viruses. 2021; 13(8):1467. https://doi.org/10.3390/v13081467
Chicago/Turabian StyleChoi, K. Yeon, and Alistair McGregor. 2021. "A Fully Protective Congenital CMV Vaccine Requires Neutralizing Antibodies to Viral Pentamer and gB Glycoprotein Complexes but a pp65 T-Cell Response Is Not Necessary" Viruses 13, no. 8: 1467. https://doi.org/10.3390/v13081467
APA StyleChoi, K. Y., & McGregor, A. (2021). A Fully Protective Congenital CMV Vaccine Requires Neutralizing Antibodies to Viral Pentamer and gB Glycoprotein Complexes but a pp65 T-Cell Response Is Not Necessary. Viruses, 13(8), 1467. https://doi.org/10.3390/v13081467