
De Novo Polycomb Recruitment: Lessons from Latent Herpesviruses


Abstract
:1. Introduction
2. Polycomb Group Protein Repressive Complexes
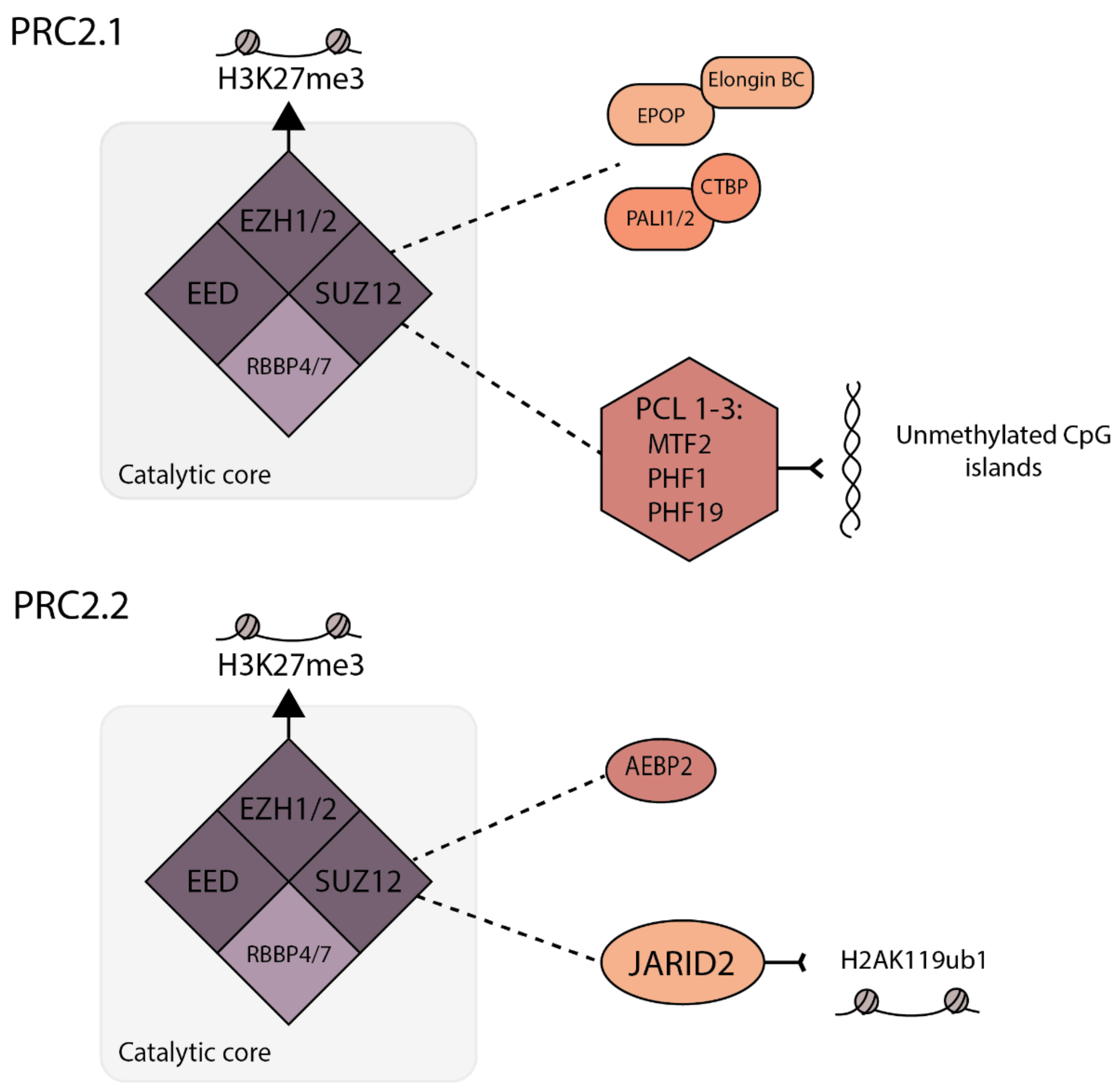
2.1. H3K27 Methylation by PRC2
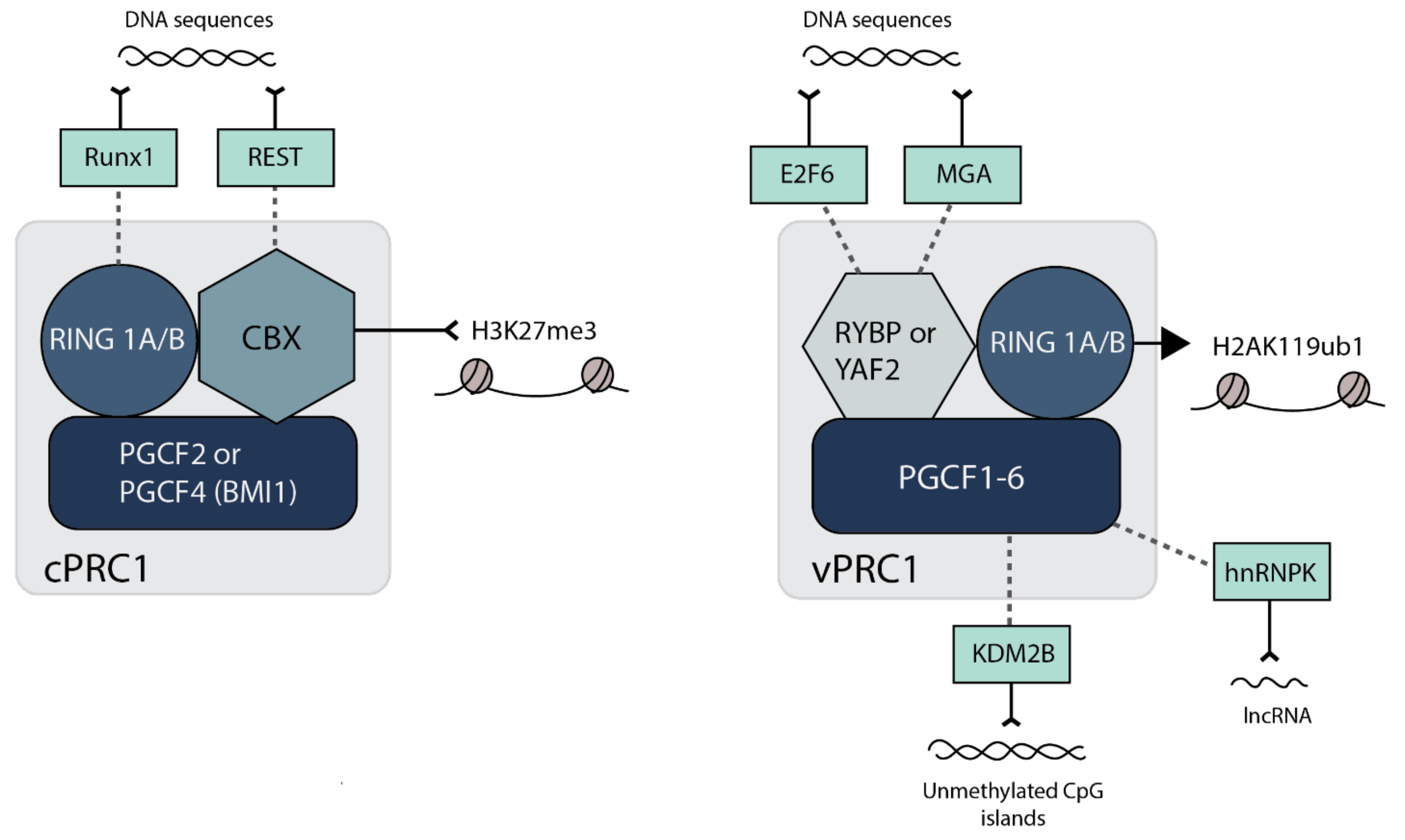
2.2. H2AK119 Ubiquitination, Chromatin Compaction and 3-Dimensional Interactions by PRC1
3. Evidence for Polycomb Group Silencing of Latent Herpesvirus Genomes
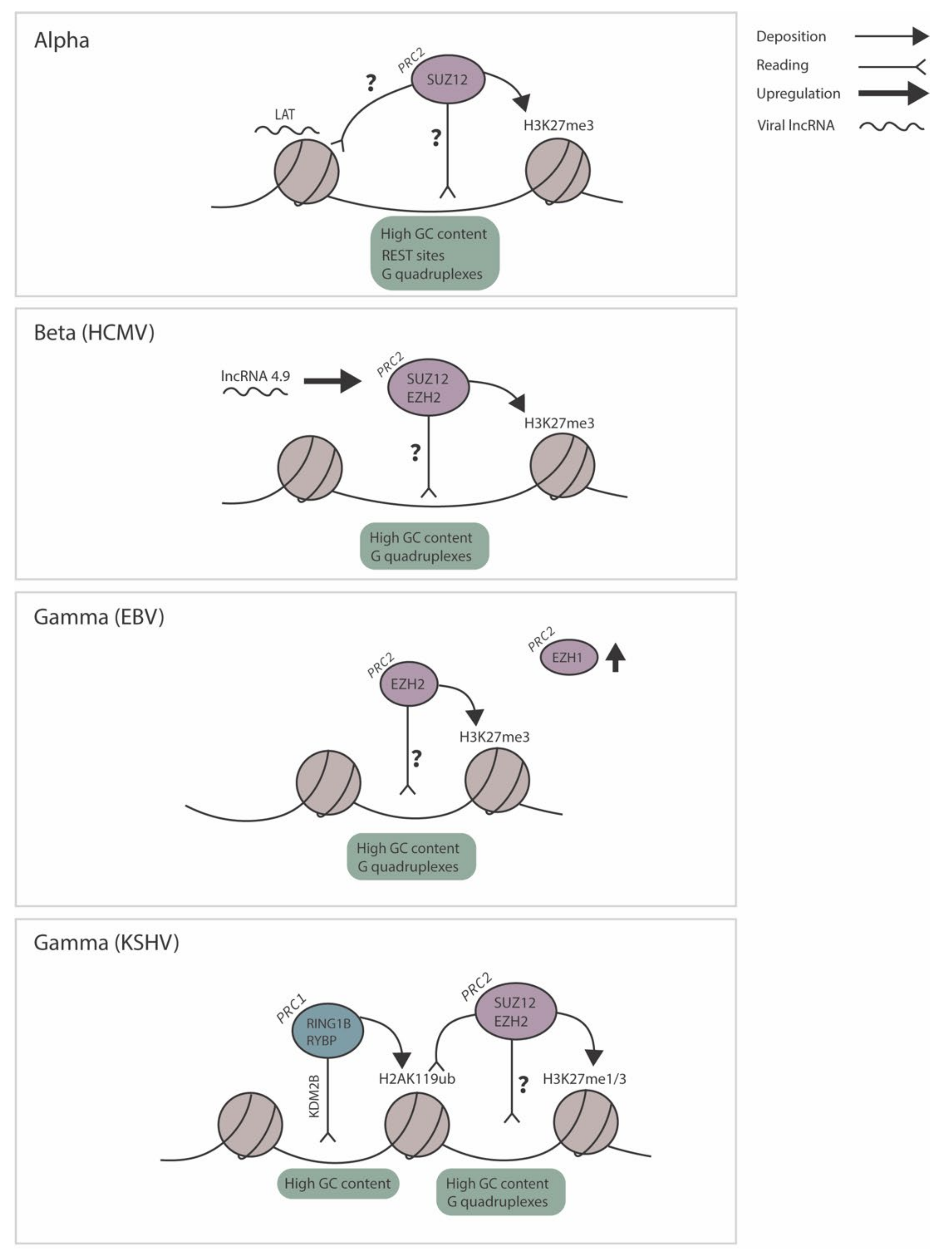
3.1. Polycomb Group Silencing of Human Alphaherpesviruses
3.2. Polycomb Group Silencing of Human Betaherpesviruses
3.3. Polycomb Group Silencing of Human Gammaherpesviruses
4. Potential Mechanisms of Polycomb Group Protein Recruitment to Herpesvirus Genomes
4.1. Kinetics of H3K27me1/2/3 Targeting to Herpesvirus Genomes
4.2. KDM2B/vPRC1-Mediated Recognition of Unmethylated CpGs
4.3. Sequence-Specific Polycomb Recruitment
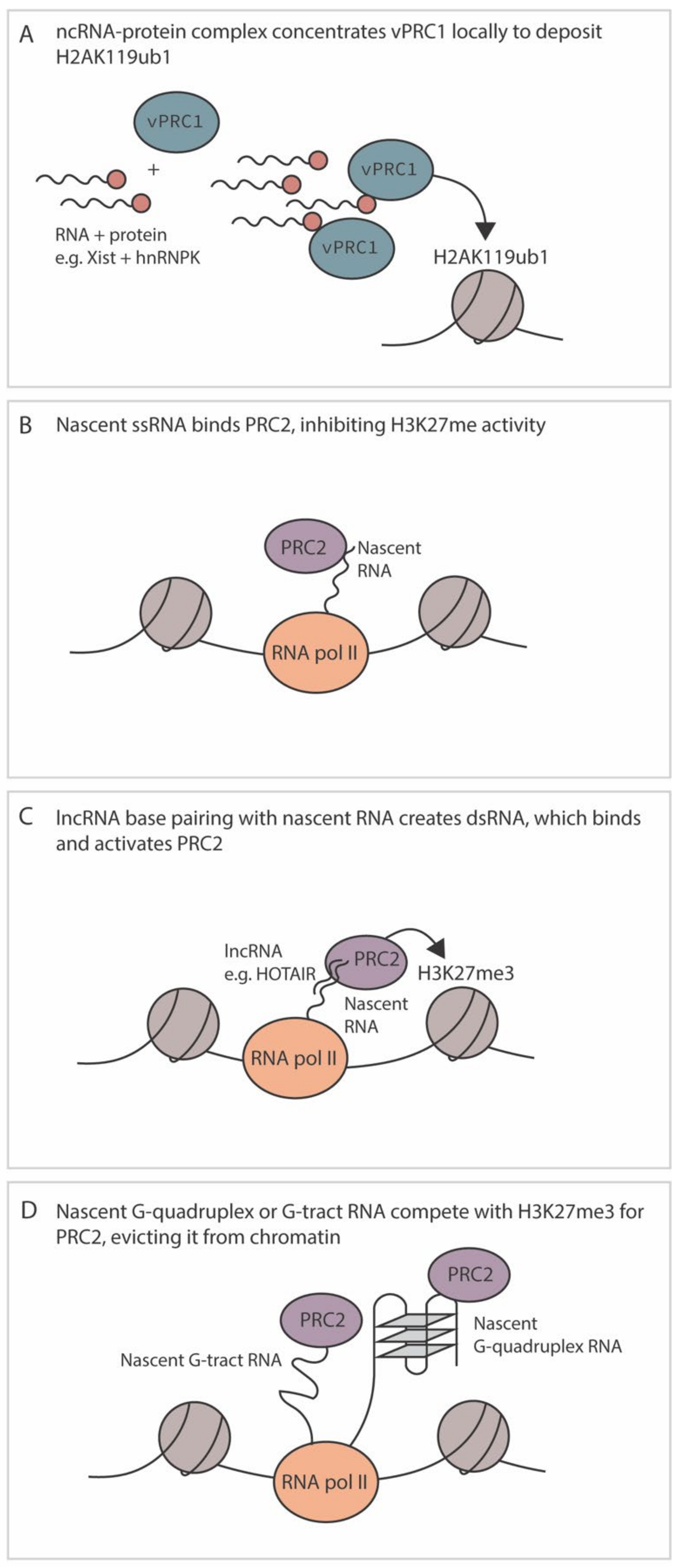
4.4. Regulation by RNA
5. Future Directions
5.1. Contribution of Cell-Type Specific PRC Complexes
5.2. Contribution of Different PRC Components to de Novo Silencing and Long-Term Repression
5.3. Viral Manipulation of the Polycomb Structure of the Virus and Heterogeneity in Latency
5.4. Mechanisms of Reversal for Reactivation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Knipe, D.M.; Cliffe, A. Chromatin control of herpes simplex virus lytic and latent infection. Nat. Rev. Microbiol. 2008, 6, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, J.; Grundhoff, A. Epigenetic control in Kaposi sarcoma-associated herpesvirus infection and associated disease. Semin. Immunopathol. 2020, 42, 143–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieberman, P.M. Chromatin Structure of Epstein–Barr Virus Latent Episomes. Curr. Top. Microbiol. Immunol. 2015, 390, 71–102. [Google Scholar] [CrossRef]
- Sinclair, J. Chromatin structure regulates human cytomegalovirus gene expression during latency, reactivation and lytic infection. Biochim. Biophys. Acta 2010, 1799, 286–295. [Google Scholar] [CrossRef]
- Trojer, P.; Reinberg, D. Facultative Heterochromatin: Is There a Distinctive Molecular Signature? Mol. Cell 2007, 28, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Loubiere, V.; Martinez, A.-M.; Cavalli, G. Cell Fate and Developmental Regulation Dynamics by Polycomb Proteins and 3D Genome Architecture. BioEssays 2019, 41, e1800222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petracovici, A.; Bonasio, R. Distinct PRC2 subunits regulate maintenance and establishment of Polycomb repression during differentiation. Mol. Cell 2021, 81, 2625–2639.e5. [Google Scholar] [CrossRef]
- Kang, S.-J.; Chun, T. Structural heterogeneity of the mammalian polycomb repressor complex in immune regulation. Exp. Mol. Med. 2020, 52, 1004–1015. [Google Scholar] [CrossRef]
- Piunti, A.; Shilatifard, A. The roles of Polycomb repressive complexes in mammalian development and cancer. Nat. Rev. Mol. Cell Biol. 2021, 22, 326–345. [Google Scholar] [CrossRef]
- Connelly, K.E.; Dykhuizen, E.C. Compositional and functional diversity of canonical PRC1 complexes in mammals. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 233–245. [Google Scholar] [CrossRef]
- Oksuz, O.; Narendra, V.; Lee, C.-H.; Descostes, N.; LeRoy, G.; Raviram, R.; Blumenberg, L.; Karch, K.; Rocha, P.; Garcia, B.A.; et al. Capturing the Onset of PRC2-Mediated Repressive Domain Formation. Mol. Cell 2018, 70, 1149–1162.e5. [Google Scholar] [CrossRef]
- Højfeldt, J.W.; Laugesen, A.; Willumsen, B.M.; Damhofer, H.; Hedehus, L.; Tvardovskiy, A.; Mohammad, F.; Jensen, O.N.; Helin, K. Accurate H3K27 methylation can be established de novo by SUZ12-directed PRC2. Nat. Struct. Mol. Biol. 2018, 25, 225–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, W.; Roizman, B. Compartmentalization of Spermine and Spermidine in the Herpes Simplex Virion. Proc. Natl. Acad. Sci. USA 1971, 68, 2818–2821. [Google Scholar] [CrossRef] [Green Version]
- Laugesen, A.; Højfeldt, J.W.; Helin, K. Molecular Mechanisms Directing PRC2 Recruitment and H3K27 Methylation. Mol. Cell 2019, 74, 8–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.-R.; Lee, C.-H.; Oksuz, O.; Stafford, J.M.; Reinberg, D. PRC2 is high maintenance. Genes Dev. 2019, 33, 903–935. [Google Scholar] [CrossRef]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of Histone H3 Lysine 27 Methylation in Polycomb-Group Silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Brown, J.; Cao, R.; Zhang, Y.; Kassis, J.; Jones, R.S. Hierarchical Recruitment of Polycomb Group Silencing Complexes. Mol. Cell 2004, 14, 637–646. [Google Scholar] [CrossRef]
- Blackledge, N.P.; Farcas, A.M.; Kondo, T.; King, H.W.; McGouran, J.F.; Hanssen, L.L.P.; Ito, S.; Cooper, S.; Kondo, K.; Koseki, Y.; et al. Variant PRC1 Complex-Dependent H2A Ubiquitylation Drives PRC2 Recruitment and Polycomb Domain Formation. Cell 2014, 157, 1445–1459. [Google Scholar] [CrossRef] [Green Version]
- Cao, R.; Zhang, Y. SUZ12 Is Required for Both the Histone Methyltransferase Activity and the Silencing Function of the EED-EZH2 Complex. Mol. Cell 2004, 15, 57–67. [Google Scholar] [CrossRef]
- Chammas, P.; Mocavini, I.; Di Croce, L. Engaging chromatin: PRC2 structure meets function. Br. J. Cancer 2020, 122, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, K.; Scelfo, A.; Jammula, S.; Cuomo, A.; Barozzi, I.; Stützer, A.; Fischle, W.; Bonaldi, T.; Pasini, D. Polycomb-Dependent H3K27me1 and H3K27me2 Regulate Active Transcription and Enhancer Fidelity. Mol. Cell 2014, 53, 49–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Mierlo, G.; Veenstra, G.J.C.; Vermeulen, M.; Marks, H. The Complexity of PRC2 Subcomplexes. Trends Cell Biol. 2019, 29, 660–671. [Google Scholar] [CrossRef] [Green Version]
- Kloet, S.L.; Makowski, M.M.; Baymaz, H.I.; van Voorthuijsen, L.; Karemaker, I.D.; Santanach, A.; Jansen, P.; Di Croce, L.; Vermeulen, M. The dynamic interactome and genomic targets of Polycomb complexes during stem-cell differentiation. Nat. Struct. Mol. Biol. 2016, 23, 682–690. [Google Scholar] [CrossRef] [Green Version]
- Beringer, M.; Pisano, P.; Di Carlo, V.; Blanco, E.; Chammas, P.; Vizan, P.; Gutiérrez, A.; Aranda, S.; Payer, B.; Wierer, M.; et al. EPOP Functionally Links Elongin and Polycomb in Pluripotent Stem Cells. Mol. Cell 2016, 64, 645–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liefke, R.; Karwacki-Neisius, V.; Shi, Y. EPOP Interacts with Elongin BC and USP7 to Modulate the Chromatin Landscape. Mol. Cell 2016, 64, 659–672. [Google Scholar] [CrossRef] [Green Version]
- Conway, E.; Jerman, E.; Healy, E.; Ito, S.; Holoch, D.; Oliviero, G.; Deevy, O.; Glancy, E.; Fitzpatrick, D.J.; Mucha, M.; et al. A Family of Vertebrate-Specific Polycombs Encoded by the LCOR/LCORL Genes Balance PRC2 Subtype Activities. Mol. Cell 2018, 70, 408–421.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, S.; Grijzenhout, A.; Underwood, E.; Ancelin, K.; Zhang, T.; Nesterova, T.B.; Anil-Kirmizitas, B.; Bassett, A.; Kooistra, S.M.; Agger, K.; et al. Jarid2 binds mono-ubiquitylated H2A lysine 119 to mediate crosstalk between Polycomb complexes PRC1 and PRC2. Nat. Commun. 2016, 7, 13661. [Google Scholar] [CrossRef] [Green Version]
- Sanulli, S.; Justin, N.; Teissandier, A.; Ancelin, K.; Portoso, M.; Caron, M.; Michaud, A.; Lombard, B.; da Rocha, S.J.T.; Offer, J.; et al. Jarid2 Methylation via the PRC2 Complex Regulates H3K27me3 Deposition during Cell Differentiation. Mol. Cell 2015, 57, 769–783. [Google Scholar] [CrossRef] [Green Version]
- Son, J.; Shen, S.; Margueron, R.; Reinberg, D. Nucleosome-binding activities within JARID2 and EZH1 regulate the function of PRC2 on chromatin. Genes Dev. 2013, 27, 2663–2677. [Google Scholar] [CrossRef] [Green Version]
- Kasinath, V.; Beck, C.; Sauer, P.; Poepsel, S.; Kosmatka, J.; Faini, M.; Toso, D.; Aebersold, R.; Nogales, E. JARID2 and AEBP2 regulate PRC2 in the presence of H2AK119ub1 and other histone modifications. Science 2021, 371, eabc3393. [Google Scholar] [CrossRef] [PubMed]
- Fursova, N.A.; Blackledge, N.P.; Nakayama, M.; Ito, S.; Koseki, Y.; Farcas, A.M.; King, H.; Koseki, H.; Klose, R.J. Synergy between Variant PRC1 Complexes Defines Polycomb-Mediated Gene Repression. Mol. Cell 2019, 74, 1020–1036.e8. [Google Scholar] [CrossRef] [Green Version]
- Youmans, D.T.; Gooding, A.R.; Dowell, R.D.; Cech, T.R. Competition between PRC2.1 and 2.2 subcomplexes regulates PRC2 chromatin occupancy in human stem cells. Mol. Cell 2021, 81, 488–501.e9. [Google Scholar] [CrossRef] [PubMed]
- Healy, E.; Mucha, M.; Glancy, E.; Fitzpatrick, D.J.; Conway, E.; Neikes, H.K.; Monger, C.; Van Mierlo, G.; Baltissen, M.P.; Koseki, Y.; et al. PRC2.1 and PRC2.2 Synergize to Coordinate H3K27 Trimethylation. Mol. Cell 2019, 76, 437–452.e6. [Google Scholar] [CrossRef]
- Hojfeldt, J.; Hedehus, L.; Laugesen, A.; Tatar, T.; Wiehle, L.; Helin, K. Non-core Subunits of the PRC2 Complex Are Collectively Required for Its Target-Site Specificity. Mol. Cell 2019, 76, 423–436.e3. [Google Scholar] [CrossRef]
- Margueron, R.; Li, G.; Sarma, K.; Blais, A.; Zavadil, J.; Woodcock, C.L.; Dynlacht, B.D.; Reinberg, D. Ezh1 and Ezh2 Maintain Repressive Chromatin through Different Mechanisms. Mol. Cell 2008, 32, 503–518. [Google Scholar] [CrossRef] [Green Version]
- Stojic, L.; Jasencakova, Z.; Prezioso, C.; Stützer, A.; Bodega, B.; Pasini, D.; Klingberg, R.; Mozzetta, C.; Margueron, R.; Puri, P.L.; et al. Chromatin regulated interchange between polycomb repressive complex 2 (PRC2)-Ezh2 and PRC2-Ezh1 complexes controls myogenin activation in skeletal muscle cells. Epigenet. Chromatin 2011, 4, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Raawi, D.; Jones, R.; Wijesinghe, S.; Halsall, J.; Petric, M.; Roberts, S.; Hotchin, N.; Kanhere, A. A novel form of JARID2 is required for differentiation in lineage-committed cells. EMBO J. 2019, 38, e98449. [Google Scholar] [CrossRef]
- Gao, Z.; Zhang, J.; Bonasio, R.; Strino, F.; Sawai, A.; Parisi, F.; Kluger, Y.; Reinberg, D. PCGF Homologs, CBX Proteins, and RYBP Define Functionally Distinct PRC1 Family Complexes. Mol. Cell 2012, 45, 344–356. [Google Scholar] [CrossRef] [Green Version]
- Fischle, W.; Wang, Y.; Jacobs, S.A.; Kim, Y.; Allis, C.D.; Khorasanizadeh, S. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. 2003, 17, 1870–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, J.; Zhang, Y.; Xu, R.-M. Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev. 2003, 17, 1823–1828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moussa, H.F.; Bsteh, D.; Yelagandula, R.; Pribitzer, C.; Stecher, K.; Bartalska, K.; Michetti, L.; Wang, J.; Zepeda-Martinez, J.A.; Elling, U.; et al. Canonical PRC1 controls sequence-independent propagation of Polycomb-mediated gene silencing. Nat. Commun. 2019, 10, 1931. [Google Scholar] [CrossRef]
- Garcia, E.; Marcos-Gutiérrez, C.; Del Mar Lorente, M.; Moreno, J.C.; Vidal, M. RYBP, a new repressor protein that interacts with components of the mammalian Polycomb complex, and with the transcription factor YY1. EMBO J. 1999, 18, 3404–3418. [Google Scholar] [CrossRef] [Green Version]
- Tavares, L.; Dimitrova, E.; Oxley, D.; Webster, J.; Poot, R.; Demmers, J.; Bezstarosti, K.; Taylor, S.; Ura, H.; Koide, H.; et al. RYBP-PRC1 Complexes Mediate H2A Ubiquitylation at Polycomb Target Sites Independently of PRC2 and H3K27me3. Cell 2012, 148, 664–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farcas, A.M.; Blackledge, N.P.; Sudbery, I.; Long, H.; McGouran, J.; Rose, N.; Lee, S.; Sims, D.; Cerase, A.; Sheahan, T.; et al. KDM2B links the Polycomb Repressive Complex 1 (PRC1) to recognition of CpG islands. eLife 2012, 1, e00205. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Kerppola, T.K. REST Interacts with Cbx Proteins and Regulates Polycomb Repressive Complex 1 Occupancy at RE1 Elements. Mol. Cell. Biol. 2011, 31, 2100–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pintacuda, G.; Wei, G.; Roustan, C.; Kirmizitas, B.A.; Solcan, N.; Cerase, A.; Castello, A.; Mohammed, S.; Moindrot, B.; Nesterova, T.; et al. hnRNPK Recruits PCGF3/5-PRC1 to the Xist RNA B-Repeat to Establish Polycomb-Mediated Chromosomal Silencing. Mol. Cell 2017, 68, 955–969.e10. [Google Scholar] [CrossRef] [Green Version]
- Lagarou, A.; Sarip, A.T.K.-M.; Moshkin, Y.; Chalkley, G.E.; Bezstarosti, K.; Demmers, J.; Verrijzer, C.P. dKDM2 couples histone H2A ubiquitylation to histone H3 demethylation during Polycomb group silencing. Genes Dev. 2008, 22, 2799–2810. [Google Scholar] [CrossRef] [Green Version]
- Blackledge, N.P.; Fursova, N.A.; Kelley, J.R.; Huseyin, M.K.; Feldmann, A.; Klose, R.J. PRC1 Catalytic Activity Is Central to Polycomb System Function. Mol. Cell 2020, 77, 857–874.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamburri, S.; Lavarone, E.; Fernández-Pérez, D.; Conway, E.; Zanotti, M.; Manganaro, D.; Pasini, D. Histone H2AK119 Mono-Ubiquitination Is Essential for Polycomb-Mediated Transcriptional Repression. Mol. Cell 2020, 77, 840–856.e5. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Zhu, P.; Wang, J.; Pascual, G.; Ohgi, K.A.; Lozach, J.; Glass, C.K.; Rosenfeld, M.G. Histone H2A Monoubiquitination Represses Transcription by Inhibiting RNA Polymerase II Transcriptional Elongation. Mol. Cell 2008, 29, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Stock, J.K.; Giadrossi, S.; Casanova, M.; Brookes, E.; Vidal, M.; Koseki, H.; Brockdorff, N.; Fisher, A.G.; Pombo, A. Ring1-mediated ubiquitination of H2A restrains poised RNA polymerase II at bivalent genes in mouse ES cells. Nat. Cell Biol. 2007, 9, 1428–1435. [Google Scholar] [CrossRef] [Green Version]
- Di Croce, L.; Helin, K. Transcriptional regulation by Polycomb group proteins. Nat. Struct. Mol. Biol. 2013, 20, 1147–1155. [Google Scholar] [CrossRef]
- Illingworth, R. Chromatin folding and nuclear architecture: PRC1 function in 3D. Curr. Opin. Genet. Dev. 2019, 55, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Morey, L.; Aloia, L.; Cozzuto, L.; Benitah, S.A.; Di Croce, L. RYBP and Cbx7 Define Specific Biological Functions of Polycomb Complexes in Mouse Embryonic Stem Cells. Cell Rep. 2013, 3, 60–69. [Google Scholar] [CrossRef] [Green Version]
- Klauke, K.; Radulović, V.; Broekhuis, M.J.C.; Weersing, E.; Zwart, E.; Olthof, S.; Ritsema, M.; Bruggeman, S.W.M.; Wu, X.; Helin, K.; et al. Polycomb Cbx family members mediate the balance between haematopoietic stem cell self-renewal and differentiation. Nat. Cell Biol. 2013, 15, 353–362. [Google Scholar] [CrossRef]
- Plys, A.J.; Davis, C.; Kim, J.; Rizki, G.; Keenen, M.M.; Marr, S.K.; Kingston, R.E. Phase separation of Polycomb-repressive complex 1 is governed by a charged disordered region of CBX2. Genes Dev. 2019, 33, 799–813. [Google Scholar] [CrossRef] [Green Version]
- Tsuboi, M.; Kishi, Y.; Yokozeki, W.; Koseki, H.; Hirabayashi, Y.; Gotoh, Y. Ubiquitination-Independent Repression of PRC1 Targets during Neuronal Fate Restriction in the Developing Mouse Neocortex. Dev. Cell 2018, 47, 758–772.e5. [Google Scholar] [CrossRef] [Green Version]
- Suzich, J.B.; Cliffe, A.R. Strength in diversity: Understanding the pathways to herpes simplex virus reactivation. Virology 2018, 522, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Sindre, H.; Tjoonnfjord, G.; Rollag, H.; Ranneberg-Nilsen, T.; Veiby, O.; Beck, S.; Degre, M.; Hestdal, K. Human cytomegalovirus suppression of and latency in early hematopoietic progenitor cells. Blood 1996, 88, 4526–4533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, G.; Jores, R.; Mocarski, E.S. Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc. Natl. Acad. Sci. USA 1998, 95, 3937–3942. [Google Scholar] [CrossRef] [Green Version]
- Elder, E.; Sinclair, J. HCMV latency: What regulates the regulators? Med. Microbiol. Immunol. 2019, 208, 431–438. [Google Scholar] [CrossRef] [Green Version]
- Dupin, N.; Fisher, C.; Kellam, P.; Ariad, S.; Tulliez, M.; Franck, N.; van Marck, E.; Salmon, D.; Gorin, I.; Escande, J.-P.; et al. Distribution of human herpesvirus-8 latently infected cells in Kaposi’s sarcoma, multicentric Castleman’s disease, and primary effusion lymphoma. Proc. Natl. Acad. Sci. USA 1999, 96, 4546–4551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bechtel, J.T.; Liang, Y.; Hvidding, J.; Ganem, D. Host Range of Kaposi’s Sarcoma-Associated Herpesvirus in Cultured Cells. J. Virol. 2003, 77, 6474–6481. [Google Scholar] [CrossRef] [Green Version]
- Boshoff, C.; Schulz, T.; Kennedy, M.M.; Graham, A.K.; Fisher, C.; Thomas, A.; McGee, J.O.; Weiss, R.A.; O’Leary, J.J. Kaposi’s sarcoma-associated herpesvirus infects endothelial and spindle cells. Nat. Med. 1995, 1, 1274–1278. [Google Scholar] [CrossRef] [PubMed]
- Thorley-Lawson, D.; Hawkins, J.; Tracy, S.; Shapiro, M. The pathogenesis of Epstein–Barr virus persistent infection. Curr. Opin. Virol. 2013, 3, 227–232. [Google Scholar] [CrossRef] [Green Version]
- Arbuckle, J.H.; Medveczky, M.M.; Luka, J.; Hadley, S.H.; Luegmayr, A.; Ablashi, D.; Lund, T.C.; Tolar, J.; De Meirleir, K.; Montoya, J.G.; et al. The latent human herpesvirus-6A genome specifically integrates in telomeres of human chromosomes in vivo and in vitro. Proc. Natl. Acad. Sci. USA 2010, 107, 5563–5568. [Google Scholar] [CrossRef] [Green Version]
- Lentine, A.F.; Bachenheimer, S.L. Intracellular organization of herpes simplex virus type 1 DNA assayed by staphylococcal nuclease sensitivity. Virus Res. 1990, 16, 275–292. [Google Scholar] [CrossRef]
- Pignatti, P.F.; Cassai, E. Analysis of herpes simplex virus nucleoprotein complexes extracted from infected cells. J. Virol. 1980, 36, 816–828. [Google Scholar] [CrossRef] [Green Version]
- Leinbach, S.S.; Summers, W.C. The Structure of Herpes Simplex Virus Type 1 DNA as Probed by Micrococcal Nuclease Digestion. J. Gen. Virol. 1980, 51, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Varnum, S.M.; Streblow, D.N.; Monroe, M.E.; Smith, P.; Auberry, K.J.; Paša-Tolić, L.; Wang, D.; Camp, D.G., 2nd; Rodland, K.; Wiley, S.; et al. Identification of Proteins in Human Cytomegalovirus (HCMV) Particles: The HCMV Proteome. J. Virol. 2004, 78, 10960–10966. [Google Scholar] [CrossRef] [Green Version]
- Johannsen, E.; Luftig, M.; Chase, M.R.; Weicksel, S.; Cahir-McFarland, E.; Illanes, D.; Sarracino, D.; Kieff, E. Proteins of purified Epstein-Barr virus. Proc. Natl. Acad. Sci. USA 2004, 101, 16286–16291. [Google Scholar] [CrossRef] [Green Version]
- Bechtel, J.T.; Winant, R.C.; Ganem, D. Host and Viral Proteins in the Virion of Kaposi’s Sarcoma-Associated Herpesvirus. J. Virol. 2005, 79, 4952–4964. [Google Scholar] [CrossRef] [Green Version]
- Reeves, M.B. Chromatin-mediated regulation of cytomegalovirus gene expression. Virus Res. 2011, 157, 134–143. [Google Scholar] [CrossRef] [Green Version]
- Buschle, A.; Hammerschmidt, W. Epigenetic lifestyle of Epstein-Barr virus. Semin. Immunopathol. 2020, 42, 131–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Q.; Seo, G.J.; Choi, Y.J.; Kwak, M.-J.; Ge, J.; Rodgers, M.; Shi, M.; Leslie, B.J.; Hopfner, K.-P.; Ha, T.; et al. Crosstalk between the cGAS DNA Sensor and Beclin-1 Autophagy Protein Shapes Innate Antimicrobial Immune Responses. Cell Host Microbe 2014, 15, 228–238. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.-H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649.e18. [Google Scholar] [CrossRef] [PubMed]
- Michalski, S.; de Oliveira Mann, C.C.; Stafford, C.A.; Witte, G.; Bartho, J.; Lammens, K.; Hornung, V.; Hopfner, K.-P. Structural basis for sequestration and autoinhibition of cGAS by chromatin. Nature 2020, 587, 678–682. [Google Scholar] [CrossRef]
- Zhao, B.; Xu, P.; Rowlett, C.M.; Jing, T.; Shinde, O.; Lei, Y.; West, A.P.; Liu, W.R.; Li, P. The molecular basis of tight nuclear tethering and inactivation of cGAS. Nature 2020, 587, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Pathare, G.R.; Decout, A.; Glück, S.; Cavadini, S.; Makasheva, K.; Hovius, R.; Kempf, G.; Weiss, J.; Kozicka, Z.; Guey, B.; et al. Structural mechanism of cGAS inhibition by the nucleosome. Nature 2020, 587, 668–672. [Google Scholar] [CrossRef] [PubMed]
- Kujirai, T.; Zierhut, C.; Takizawa, Y.; Kim, R.; Negishi, L.; Uruma, N.; Hirai, S.; Funabiki, H.; Kurumizaka, H. Structural basis for the inhibition of cGAS by nucleosomes. Science 2020, 370, 455–458. [Google Scholar] [CrossRef]
- Boyer, J.A.; Spangler, C.J.; Strauss, J.D.; Cesmat, A.P.; Liu, P.; McGinty, R.K.; Zhang, Q. Structural basis of nucleosome-dependent cGAS inhibition. Science 2020, 370, 450–454. [Google Scholar] [CrossRef]
- Cliffe, A.R.; Garber, D.A.; Knipe, D.M. Transcription of the Herpes Simplex Virus Latency-Associated Transcript Promotes the Formation of Facultative Heterochromatin on Lytic Promoters. J. Virol. 2009, 83, 8182–8190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.-Y.; Zhou, C.; Johnson, K.E.; Colgrove, R.C.; Coen, D.M.; Knipe, D.M. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc. Natl. Acad. Sci. USA 2005, 102, 16055–16059. [Google Scholar] [CrossRef] [Green Version]
- Kwiatkowski, D.L.; Thompson, H.W.; Bloom, D.C. The Polycomb Group Protein Bmi1 Binds to the Herpes Simplex Virus 1 Latent Genome and Maintains Repressive Histone Marks during Latency. J. Virol. 2009, 83, 8173–8181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, H.C.; Lu, J.; Verma, S.C.; Banerjee, S.; Mehta, D.; Robertson, E.S. Kaposi’s Sarcoma-Associated Herpesvirus Genome Programming during the Early Stages of Primary Infection of Peripheral Blood Mononuclear Cells. mBio 2014, 5, e02261-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, R.; Tan, X.; Wang, X.; Wang, X.; Yang, L.; Robertson, E.S.; Lan, K. Epigenetic Landscape of Kaposi’s Sarcoma-Associated Herpesvirus Genome in Classic Kaposi’s Sarcoma Tissues. PLoS Pathog. 2017, 13, e1006167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeves, M.; MacAry, P.A.; Lehner, P.J.; Sissons, J.G.P.; Sinclair, J.H. Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc. Natl. Acad. Sci. USA 2005, 102, 4140–4145. [Google Scholar] [CrossRef] [Green Version]
- Reeves, M.B.; Sinclair, J.H. Circulating Dendritic Cells Isolated from Healthy Seropositive Donors Are Sites of Human Cytomegalovirus Reactivation In Vivo. J. Virol. 2013, 87, 10660–10667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Günther, T.; Grundhoff, A. The Epigenetic Landscape of Latent Kaposi Sarcoma-Associated Herpesvirus Genomes. PLoS Pathog. 2010, 6, e1000935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toth, Z.; Maglinte, D.T.; Lee, S.H.; Lee, H.-R.; Wong, L.-Y.; Brulois, K.F.; Lee, S.; Buckley, J.D.; Laird, P.W.; Marquez, V.E.; et al. Epigenetic Analysis of KSHV Latent and Lytic Genomes. PLoS Pathog. 2010, 6, e1001013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramasubramanyan, S.; Osborn, K.; Flower, K.; Sinclair, A.J. Dynamic Chromatin Environment of Key Lytic Cycle Regulatory Regions of the Epstein-Barr Virus Genome. J. Virol. 2011, 86, 1809–1819. [Google Scholar] [CrossRef] [Green Version]
- Murata, T.; Kondo, Y.; Sugimoto, A.; Kawashima, D.; Saito, S.; Isomura, H.; Kanda, T.; Tsurumi, T. Epigenetic Histone Modification of Epstein-Barr Virus BZLF1 Promoter during Latency and Reactivation in Raji Cells. J. Virol. 2012, 86, 4752–4761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubat, N.J.; Tran, R.K.; McAnany, P.; Bloom, D.C. Specific Histone Tail Modification and Not DNA Methylation Is a Determinant of Herpes Simplex Virus Type 1 Latent Gene Expression. J. Virol. 2004, 78, 1139–1149. [Google Scholar] [CrossRef] [Green Version]
- Kubat, N.J.; Amelio, A.L.; Giordani, N.V.; Bloom, D.C. The Herpes Simplex Virus Type 1 Latency-Associated Transcript (LAT) Enhancer/ rcr Is Hyperacetylated during Latency Independently of LAT Transcription. J. Virol. 2004, 78, 12508–12518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cliffe, A.R.; Coen, D.M.; Knipe, D.M. Kinetics of Facultative Heterochromatin and Polycomb Group Protein Association with the Herpes Simplex Viral Genome during Establishment of Latent Infection. mBio 2013, 4, e00590-12. [Google Scholar] [CrossRef] [Green Version]
- Marques, S.; Efstathiou, S.; Smith, K.G.; Haury, M.; Simas, J.P. Selective Gene Expression of Latent Murine Gammaherpesvirus 68 in B Lymphocytes. J. Virol. 2003, 77, 7308–7318. [Google Scholar] [CrossRef] [Green Version]
- Cliffe, A.R.; Arbuckle, J.H.; Vogel, J.L.; Geden, M.J.; Rothbart, S.B.; Cusack, C.L.; Strahl, B.D.; Kristie, T.M.; Deshmukh, M. Neuronal Stress Pathway Mediating a Histone Methyl/Phospho Switch Is Required for Herpes Simplex Virus Reactivation. Cell Host Microbe 2015, 18, 649–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossetto, C.C.; Tarrant-Elorza, M.; Pari, G.S. Cis and Trans Acting Factors Involved in Human Cytomegalovirus Experimental and Natural Latent Infection of CD14 (+) Monocytes and CD34 (+) Cells. PLoS Pathog. 2013, 9, e1003366. [Google Scholar] [CrossRef] [Green Version]
- Thellman, N.M.; Botting, C.; Madaj, Z.; Triezenberg, S.J. An Immortalized Human Dorsal Root Ganglion Cell Line Provides a Novel Context To Study Herpes Simplex Virus 1 Latency and Reactivation. J. Virol. 2017, 91, e00080-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, T.G.; Bloom, D.C. Lund Human Mesencephalic (LUHMES) Neuronal Cell Line Supports Herpes Simplex Virus 1 Latency In Vitro. J. Virol. 2019, 93, 02210–02218. [Google Scholar] [CrossRef] [Green Version]
- Henriquez, B.; Bustos, F.; Aguilar, R.; Becerra, A.; Simon, F.; Montecino, M.; Van Zundert, B. Ezh1 and Ezh2 differentially regulate PSD-95 gene transcription in developing hippocampal neurons. Mol. Cell. Neurosci. 2013, 57, 130–143. [Google Scholar] [CrossRef]
- Von Schimmelmann, M.; Feinberg, P.; Sullivan, J.M.; Ku, S.M.; Badimon, A.; Duff, M.K.; Wang, Z.; Lachmann, A.; Dewell, S.; Ma’Ayan, A.; et al. Polycomb repressive complex 2 (PRC2) silences genes responsible for neurodegeneration. Nat. Neurosci. 2016, 19, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, M.P.; Hann, W.; Shivkumar, M.; Harman, L.E.R.; Connor, V.; Coleman, H.M.; Proença, J.; Efstathiou, S. The HSV-1 Latency-Associated Transcript Functions to Repress Latent Phase Lytic Gene Expression and Suppress Virus Reactivation from Latently Infected Neurons. PLoS Pathog. 2016, 12, e1005539. [Google Scholar] [CrossRef] [PubMed]
- Hancock, M.; Skalsky, R.L. Roles of Non-coding RNAs during Herpesvirus Infection. Curr. Top. Microbiol. Immunol. 2018, 419, 243–280. [Google Scholar] [CrossRef]
- Messer, H.G.P.; Jacobs, D.; Dhummakupt, A.; Bloom, D.C.; Utt, A.; Das, P.K.; Varjak, M.; Lulla, V.; Lulla, A.; Merits, A. Inhibition of H3K27me3-Specific Histone Demethylases JMJD3 and UTX Blocks Reactivation of Herpes Simplex Virus 1 in Trigeminal Ganglion Neurons. J. Virol. 2015, 89, 3417–3420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertke, A.S.; Patel, A.; Krause, P.R. Herpes Simplex Virus Latency-Associated Transcript Sequence Downstream of the Promoter Influences Type-Specific Reactivation and Viral Neurotropism. J. Virol. 2007, 81, 6605–6613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanez, A.A.; Harrell, T.; Sriranganathan, H.J.; Ives, A.M.; Bertke, A.S. Neurotrophic Factors NGF, GDNF and NTN Selectively Modulate HSV1 and HSV2 Lytic Infection and Reactivation in Primary Adult Sensory and Autonomic Neurons. Pathogens 2017, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Dukhovny, A.; Sloutskin, A.; Markus, A.; Yee, M.; Kinchington, P.R.; Goldstein, R.S. Varicella-Zoster Virus Infects Human Embryonic Stem Cell-Derived Neurons and Neurospheres but Not Pluripotent Embryonic Stem Cells or Early Progenitors. J. Virol. 2012, 86, 3211–3218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laemmle, L.; Goldstein, R.S.; Kinchington, P.R. Modeling Varicella Zoster Virus Persistence and Reactivation – Closer to Resolving a Perplexing Persistent State. Front. Microbiol. 2019, 10, 1634. [Google Scholar] [CrossRef] [Green Version]
- Abraham, C.G.; Kulesza, C.A. Polycomb Repressive Complex 2 Silences Human Cytomegalovirus Transcription in Quiescent Infection Models. J. Virol. 2013, 87, 13193–13205. [Google Scholar] [CrossRef] [Green Version]
- Gan, X.; Wang, H.; Yu, Y.; Yi, W.; Zhu, S.; Li, E.; Liang, Y. Epigenetically repressing human cytomegalovirus lytic infection and reactivation from latency in THP-1 model by targeting H3K9 and H3K27 histone demethylases. PLoS ONE 2017, 12, e0175390. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Albright, E.R.; Lee, J.-H.; Jacobs, D.; Kalejta, R.F. Cellular defense against latent colonization foiled by human cytomegalovirus UL138 protein. Sci. Adv. 2015, 1, e1501164. [Google Scholar] [CrossRef]
- Reeves, M.; Lehner, P.J.; Sissons, J.G.P.; Sinclair, J.H. An in vitro model for the regulation of human cytomegalovirus latency and reactivation in dendritic cells by chromatin remodelling. J. Gen. Virol. 2005, 86, 2949–2954. [Google Scholar] [CrossRef]
- Kew, V.G.; Yuan, J.; Meier, J.; Reeves, M.B. Mitogen and Stress Activated Kinases Act Co-operatively with CREB during the Induction of Human Cytomegalovirus Immediate-Early Gene Expression from Latency. PLoS Pathog. 2014, 10, e1004195. [Google Scholar] [CrossRef] [PubMed]
- Strenger, V.; Caselli, E.; Lautenschlager, I.; Schwinger, W.; Aberle, S.W.; Loginov, R.; Gentili, V.; Nacheva, E.; Di Luca, D.; Urban, C. Detection of HHV-6-specific mRNA and antigens in PBMCs of individuals with chromosomally integrated HHV-6 (ciHHV-6). Clin. Microbiol. Infect. 2014, 20, 1027–1032. [Google Scholar] [CrossRef] [Green Version]
- Saviola, A.J.; Zimmermann, C.; Mariani, M.P.; Signorelli, S.A.; Gerrard, D.L.; Boyd, J.R.; Wight, D.J.; Morissette, G.; Gravel, A.; Dubuc, I.; et al. Chromatin Profiles of Chromosomally Integrated Human Herpesvirus-6A. Front. Microbiol. 2019, 10, 1408. [Google Scholar] [CrossRef] [PubMed]
- Toth, Z.; Brulois, K.; Lee, H.-R.; Izumiya, Y.; Tepper, C.; Kung, H.-J.; Jung, J.U. Biphasic Euchromatin-to-Heterochromatin Transition on the KSHV Genome Following De Novo Infection. PLoS Pathog. 2013, 9, e1003813. [Google Scholar] [CrossRef] [PubMed]
- Stürzl, M.; Gaus, D.; Dirks, W.G.; Ganem, N.; Jochmann, R. Kaposi’s sarcoma-derived cell line SLK is not of endothelial origin, but is a contaminant from a known renal carcinoma cell line. Int. J. Cancer 2013, 132, 1954–1958. [Google Scholar] [CrossRef] [PubMed]
- Toth, Z.; Papp, B.; Brulois, K.; Choi, Y.J.; Gao, S.-J.; Jung, J.U. LANA-Mediated Recruitment of Host Polycomb Repressive Complexes onto the KSHV Genome during De Novo Infection. PLoS Pathog. 2016, 12, e1005878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Günther, T.; Fröhlich, J.; Herrde, C.; Ohno, S.; Burkhardt, L.; Adler, H.; Grundhoff, A. A comparative epigenome analysis of gammaherpesviruses suggests cis-acting sequence features as critical mediators of rapid polycomb recruitment. PLoS Pathog. 2019, 15, e1007838. [Google Scholar] [CrossRef] [Green Version]
- Naik, N.G.; Nguyen, T.H.; Roberts, L.; Fischer, L.T.; Glickman, K.; Golas, G.; Papp, B.; Toth, Z. Epigenetic factor siRNA screen during primary KSHV infection identifies novel host restriction factors for the lytic cycle of KSHV. PLoS Pathog. 2020, 16, e1008268. [Google Scholar] [CrossRef] [PubMed]
- Hopcraft, S.E.; Pattenden, S.G.; James, L.I.; Frye, S.; Dittmer, D.P.; Damania, B. Chromatin remodeling controls Kaposi’s sarcoma-associated herpesvirus reactivation from latency. PLoS Pathog. 2018, 14, e1007267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, L.S.; Rickinson, A.B. Epstein–Barr virus: 40 years on. Nat. Rev. Cancer 2004, 4, 757–768. [Google Scholar] [CrossRef]
- Münz, C. Latency and lytic replication in Epstein–Barr virus-associated oncogenesis. Nat. Rev. Microbiol. 2019, 17, 691–700. [Google Scholar] [CrossRef] [Green Version]
- Tempera, I.; Lieberman, P.M. Epigenetic regulation of EBV persistence and oncogenesis. Semin. Cancer Biol. 2014, 26, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Woellmer, A.; Arteaga-Salas, J.M.; Hammerschmidt, W. BZLF1 Governs CpG-Methylated Chromatin of Epstein-Barr Virus Reversing Epigenetic Repression. PLoS Pathog. 2012, 8, e1002902. [Google Scholar] [CrossRef]
- Vargas-Ayala, R.C.; Jay, A.; Manara, F.; Maroui, M.A.; Hernandez-Vargas, H.; Diederichs, A.; Robitaille, A.; Sirand, C.; Ceraolo, M.G.; Romero-Medina, M.C.; et al. Interplay between the Epigenetic Enzyme Lysine (K)-Specific Demethylase 2B and Epstein-Barr Virus Infection. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaeffner, M.; Mrozek-Gorska, P.; Buschle, A.; Woellmer, A.; Tagawa, T.; Cernilogar, F.M.; Schotta, G.; Krietenstein, N.; Lieleg, C.; Korber, P.; et al. BZLF1 interacts with chromatin remodelers promoting escape from latent infections with EBV. Life Sci. Alliance 2019, 2, e201800108. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, T.; Okuno, Y.; Sato, Y.; Goshima, F.; Yoshiyama, H.; Kanda, T.; Kimura, H.; Murata, T. Regulation of Epstein-Barr Virus Life Cycle and Cell Proliferation by Histone H3K27 Methyltransferase EZH2 in Akata Cells. mSphere 2018, 3, e00478-18. [Google Scholar] [CrossRef] [Green Version]
- Imai, K.; Kamio, N.; Cueno, M.E.; Saito, Y.; Inoue, H.; Saito, I.; Ochiai, K. Role of the histone H3 lysine 9 methyltransferase Suv39 h1 in maintaining Epsteinn-Barr virus latency in B95-8 cells. FEBS J. 2014, 281, 2148–2158. [Google Scholar] [CrossRef] [PubMed]
- Günther, T.; Schreiner, S.; Dobner, T.; Tessmer, U.; Grundhoff, A. Influence of ND10 Components on Epigenetic Determinants of Early KSHV Latency Establishment. PLoS Pathog. 2014, 10, e1004274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrozek-Gorska, P.; Buschle, A.; Pich, D.; Schwarzmayr, T.; Fechtner, R.; Scialdone, A.; Hammerschmidt, W. Epstein–Barr virus reprograms human B lymphocytes immediately in the prelatent phase of infection. Proc. Natl. Acad. Sci. USA 2019, 116, 16046–16055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalla, M.; Schmeinck, A.; Bergbauer, M.; Pich, D.; Hammerschmidt, W. AP-1 homolog BZLF1 of Epstein-Barr virus has two essential functions dependent on the epigenetic state of the viral genome. Proc. Natl. Acad. Sci. USA 2010, 107, 850–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.; Liu, W.; Yi, X.; Yang, Y.; Su, D.; Huang, W.; Yu, H.; Teng, X.; Yang, Y.; Feng, W.; et al. PHF20L1 as a H3K27me2 reader coordinates with transcriptional repressors to promote breast tumorigenesis. Sci. Adv. 2020, 6, eaaz0356. [Google Scholar] [CrossRef] [Green Version]
- Poepsel, S.; Kasinath, V.; Nogales, E. Cryo-EM structures of PRC2 simultaneously engaged with two functionally distinct nucleosomes. Nat. Struct. Mol. Biol. 2018, 25, 154–162. [Google Scholar] [CrossRef]
- Albright, E.R.; Kalejta, R.F. Canonical and Variant Forms of Histone H3 Are Deposited onto the Human Cytomegalovirus Genome during Lytic and Latent Infections. J. Virol. 2016, 90, 10309–10320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlke, C.; Maul, K.; Christalla, T.; Walz, N.; Schult, P.; Stocking, C.; Grundhoff, A. A microRNA Encoded by Kaposi Sarcoma-Associated Herpesvirus Promotes B-Cell Expansion In Vivo. PLoS ONE 2012, 7, e49435. [Google Scholar] [CrossRef] [Green Version]
- Wood, C.D.; Carvell, T.; Gunnell, A.; Ojeniyi, O.O.; Osborne, C.; West, M.J. Enhancer Control of MicroRNA miR-155 Expression in Epstein-Barr Virus-Infected B Cells. J. Virol. 2018, 92, e00716-18. [Google Scholar] [CrossRef] [Green Version]
- Stielow, B.; Finkernagel, F.; Stiewe, T.; Nist, A.; Suske, G. MGA, L3MBTL2 and E2F6 determine genomic binding of the non-canonical Polycomb repressive complex PRC1.6. PLoS Genet. 2018, 14, e1007193. [Google Scholar] [CrossRef]
- Yu, M.; Mazor, T.; Huang, H.; Huang, H.-T.; Kathrein, K.L.; Woo, A.; Chouinard, C.R.; Labadorf, A.; Akie, T.E.; Moran, T.B.; et al. Direct Recruitment of Polycomb Repressive Complex 1 to Chromatin by Core Binding Transcription Factors. Mol. Cell 2012, 45, 330–343. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, N.; Lerdrup, M.; Landt, E.M.; Agrawal-Singh, S.; Bak, M.; Tommerup, N.; Rappsilber, J.; Södersten, E.; Hansen, K. REST–Mediated Recruitment of Polycomb Repressor Complexes in Mammalian Cells. PLoS Genet. 2012, 8, e1002494. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.J.; Khoury-Hanold, W.; Jain, P.C.; Klein, J.; Kong, Y.; Pope, S.D.; Ge, W.; Medzhitov, R.; Iwasaki, A. RUNX Binding Sites Are Enriched in Herpesvirus Genomes, and RUNX1 Overexpression Leads to Herpes Simplex Virus 1 Suppression. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- West, M.J.; Farrell, P.J. Roles of RUNX in B Cell Immortalisation. Adv. Exp. Med. Biol. 2017, 962, 283–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, T.; Zhou, G.; Khan, S.; Gu, H.; Roizman, B. Disruption of HDAC/CoREST/REST repressor by dnREST reduces genome silencing and increases virulence of herpes simplex virus. Proc. Natl. Acad. Sci. USA 2010, 107, 15904–15909. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Du, T.; Roizman, B. HSV carrying WT REST establishes latency but reactivates only if the synthesis of REST is suppressed. Proc. Natl. Acad. Sci. USA 2013, 110, E498–E506. [Google Scholar] [CrossRef] [Green Version]
- Long, Y.; Hwang, T.; Gooding, A.R.; Goodrich, K.J.; Rinn, J.L.; Cech, T.R. RNA is essential for PRC2 chromatin occupancy and function in human pluripotent stem cells. Nat. Genet. 2020, 52, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.; Bowness, J.S.; Brockdorff, N. The many faces of Polycomb regulation by RNA. Curr. Opin. Genet. Dev. 2020, 61, 53–61. [Google Scholar] [CrossRef]
- Davidovich, C.; Cech, T.R. The recruitment of chromatin modifiers by long noncoding RNAs: Lessons from PRC2. RNA 2015, 21, 2007–2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trotman, J.B.; Braceros, K.C.A.; Cherney, R.E.; Murvin, M.M.; Calabrese, J.M. The control of polycomb repressive complexes by long noncoding RNAs. Wiley Interdiscip. Rev. RNA 2021, e1657. [Google Scholar] [CrossRef]
- Zhao, J.; Sun, B.; Erwin, J.; Song, J.-J.; Lee, J.T. Polycomb Proteins Targeted by a Short Repeat RNA to the Mouse X Chromosome. Science 2008, 322, 750–756. [Google Scholar] [CrossRef] [Green Version]
- Beltran, M.; Tavares, M.; Justin, N.; Khandelwal, G.; Ambrose, J.; Foster, B.; Worlock, K.B.; Tvardovskiy, A.; Kunzelmann, S.; Herrero, J.; et al. G-tract RNA removes Polycomb repressive complex 2 from genes. Nat. Struct. Mol. Biol. 2019, 26, 899–909. [Google Scholar] [CrossRef]
- Balas, M.M.; Hartwick, E.W.; Barrington, C.; Roberts, J.T.; Wu, S.K.; Bettcher, R.; Griffin, A.M.; Kieft, J.S.; Johnson, A.M. Establishing RNA-RNA interactions remodels lncRNA structure and promotes PRC2 activity. Sci. Adv. 2021, 7, eabc9191. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; e Silva, M.S.; Jaber, T.; Vitvitskaia, O.; Li, S.; Henderson, G.; Jones, C. Two Small RNAs Encoded within the First 1.5 Kilobases of the Herpes Simplex Virus Type 1 Latency-Associated Transcript Can Inhibit Productive Infection and Cooperate to Inhibit Apoptosis. J. Virol. 2009, 83, 9131–9139. [Google Scholar] [CrossRef] [Green Version]
- Garber, D.; Schaffer, P.; Knipe, D.M. A LAT-associated function reduces productive-cycle gene expression during acute infection of murine sensory neurons with herpes simplex virus type 1. J. Virol. 1997, 71, 5885–5893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.H.; Kramer, M.F.; Schaffer, P.; Coen, D.M. A viral function represses accumulation of transcripts from productive-cycle genes in mouse ganglia latently infected with herpes simplex virus. J. Virol. 1997, 71, 5878–5884. [Google Scholar] [CrossRef] [Green Version]
- Vanni, E.A.H.; Foley, J.W.; Davison, A.J.; Sommer, M.; Liu, D.; Sung, P.; Moffat, J.; Zerboni, L.; Arvin, A.M. The latency-associated transcript locus of herpes simplex virus 1 is a virulence determinant in human skin. PLoS Pathog. 2020, 16, e1009166. [Google Scholar] [CrossRef]
- Perng, G.C.; Jones, C.; Ciacci-Zanella, J.; Stone, M.; Henderson, G.; Yukht, A.; Slanina, S.M.; Hofman, F.M.; Ghiasi, H.; Nesburn, A.B.; et al. Virus-Induced Neuronal Apoptosis Blocked by the Herpes Simplex Virus Latency-Associated Transcript. Science 2000, 287, 1500–1503. [Google Scholar] [CrossRef] [Green Version]
- Thompson, R.L.; Sawtell, N.M. Herpes Simplex Virus Type 1 Latency-Associated Transcript Gene Promotes Neuronal Survival. J. Virol. 2001, 75, 6660–6675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frasson, I.; Nadai, M.; Richter, S.N. Conserved G-Quadruplexes Regulate the Immediate Early Promoters of Human Alphaherpesviruses. Molecules 2019, 24, 2375. [Google Scholar] [CrossRef] [Green Version]
- Artusi, S.; Nadai, M.; Perrone, R.; Biasolo, M.A.; Palù, G.; Flamand, L.; Calistri, A.; Richter, S.N. The Herpes Simplex Virus-1 genome contains multiple clusters of repeated G-quadruplex: Implications for the antiviral activity of a G-quadruplex ligand. Antivir. Res. 2015, 118, 123–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, B.; Kandpal, M.; Jauhari, U.K.; Vivekanandan, P. Genome-wide analysis of G-quadruplexes in herpesvirus genomes. BMC Genom. 2016, 17, 949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossetto, C.C.; Tarrant-Elorza, M.; Verma, S.C.; Purushothaman, P.; Pari, G.S. Regulation of Viral and Cellular Gene Expression by Kaposi’s Sarcoma-Associated Herpesvirus Polyadenylated Nuclear RNA. J. Virol. 2013, 87, 5540–5553. [Google Scholar] [CrossRef] [Green Version]
- Withers, J.B.; Li, E.S.; Vallery, T.K.; Yario, T.A.; Steitz, J.A. Two herpesviral noncoding PAN RNAs are functionally homologous but do not associate with common chromatin loci. PLoS Pathog. 2018, 14, e1007389. [Google Scholar] [CrossRef] [PubMed]
- Cuddy, S.R.; Schinlever, A.R.; Dochnal, S.; Seegren, P.V.; Suzich, J.; Kundu, P.; Downs, T.K.; Farah, M.; Desai, B.N.; Boutell, C.; et al. Neuronal hyperexcitability is a DLK-dependent trigger of herpes simplex virus reactivation that can be induced by IL-1. eLife 2020, 9. [Google Scholar] [CrossRef]
- Gehani, S.S.; Agrawal-Singh, S.; Dietrich, N.; Christophersen, N.S.; Helin, K.; Hansen, K. Polycomb Group Protein Displacement and Gene Activation through MSK-Dependent H3K27me3S28 Phosphorylation. Mol. Cell 2010, 39, 886–900. [Google Scholar] [CrossRef] [PubMed]
- Lau, P.N.I.; Cheung, P. Histone code pathway involving H3 S28 phosphorylation and K27 acetylation activates transcription and antagonizes polycomb silencing. Proc. Natl. Acad. Sci. USA 2011, 108, 2801–2806. [Google Scholar] [CrossRef] [Green Version]
- Palomer, E.; Carretero, J.; Benvegnù, S.; Dotti, C.G.; Martin, M.G. Neuronal activity controls Bdnf expression via Polycomb de-repression and CREB/CBP/JMJD3 activation in mature neurons. Nat. Commun. 2016, 7, 11081. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Full-Length Name | Abbreviation |
|---|---|---|
| Polycomb repressive complex 1 | PRC1 | |
| Ring Finger Protein 1A, B | RING1A, B | |
| Polycomb group RING finger protein | PCGF | |
| Canonical PRC1 | cPRC1 | |
| Polycomb group RING finger protein 2, melanoma nuclear protein 18 | PCGF2/MEL18 | |
| Polycomb group RING finger protein 4 | PCGF4/BMI1 | |
| Chromobox 2, 4, 6, 7, 8 | CBX2, 4, 6, 7, 8 | |
| * Runt-related transcription factor 1 | RUNX1 | |
| * RE1-silencing transcription factor/ neuron-restrictive silencing factor | REST/NRSF | |
| Non-canonical/variant PRC1 | vPRC1 | |
| RING1 and YY1-binding protein | RYBP | |
| YY1-associated factor 2 | YAF2 | |
| Lysine (K)-specific demethylase 2B | KDM2B | |
| Polycomb group RING finger protein 1–6 | PCGF 1–6 | |
| * E2F transcription factor 6 | E2F6 | |
| * MAX gene-associated protein | MGA | |
| * Heterogeneous nuclear ribonucleoprotein K | hnRNPK | |
| Polycomb Repressive complex 2 | PRC2 | |
| Enhancer of Zeste 1, 2 | EZH1, 2 | |
| Suppressor of Zeste 12 | SUZ12 | |
| Embryonic ectoderm development | EED | |
| Retinoblastoma-associated proteins 46 | RbAp46/RBBP4 | |
| Retinoblastoma-associated proteins 48 | RbAp48/RBBP7 | |
| PRC2.1 | ||
| Elongin BC | No abbreviation | |
| Elongin BC- and PRC2-associated Protein | EPOP | |
| PRC2-associated LCOR isoform1, 2 C-terminal binding protein | PAL1, 2 CTBP | |
| Polycomb-like protein 1/PHD finger protein 1 | PCL1/PHF1 | |
| Polycomb-like protein 2/metal response element binding transcription factor 2 | PCL2/MTF2 | |
| Polycomb-like protein 3/PHD finger protein 19 | PCL3/PHF19 | |
| PRC2.2 | ||
| Jumonji and AT-rich interaction domain 2 | JARID2 | |
| Adipocyte enhancer-binding protein 2 | AEBP2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dochnal, S.A.; Francois, A.K.; Cliffe, A.R. De Novo Polycomb Recruitment: Lessons from Latent Herpesviruses. Viruses 2021, 13, 1470. https://doi.org/10.3390/v13081470
Dochnal SA, Francois AK, Cliffe AR. De Novo Polycomb Recruitment: Lessons from Latent Herpesviruses. Viruses. 2021; 13(8):1470. https://doi.org/10.3390/v13081470
Chicago/Turabian StyleDochnal, Sara A., Alison K. Francois, and Anna R. Cliffe. 2021. "De Novo Polycomb Recruitment: Lessons from Latent Herpesviruses" Viruses 13, no. 8: 1470. https://doi.org/10.3390/v13081470
APA StyleDochnal, S. A., Francois, A. K., & Cliffe, A. R. (2021). De Novo Polycomb Recruitment: Lessons from Latent Herpesviruses. Viruses, 13(8), 1470. https://doi.org/10.3390/v13081470