
Buprenorphine Increases HIV-1 Infection In Vitro but Does Not Reactivate HIV-1 from Latency

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
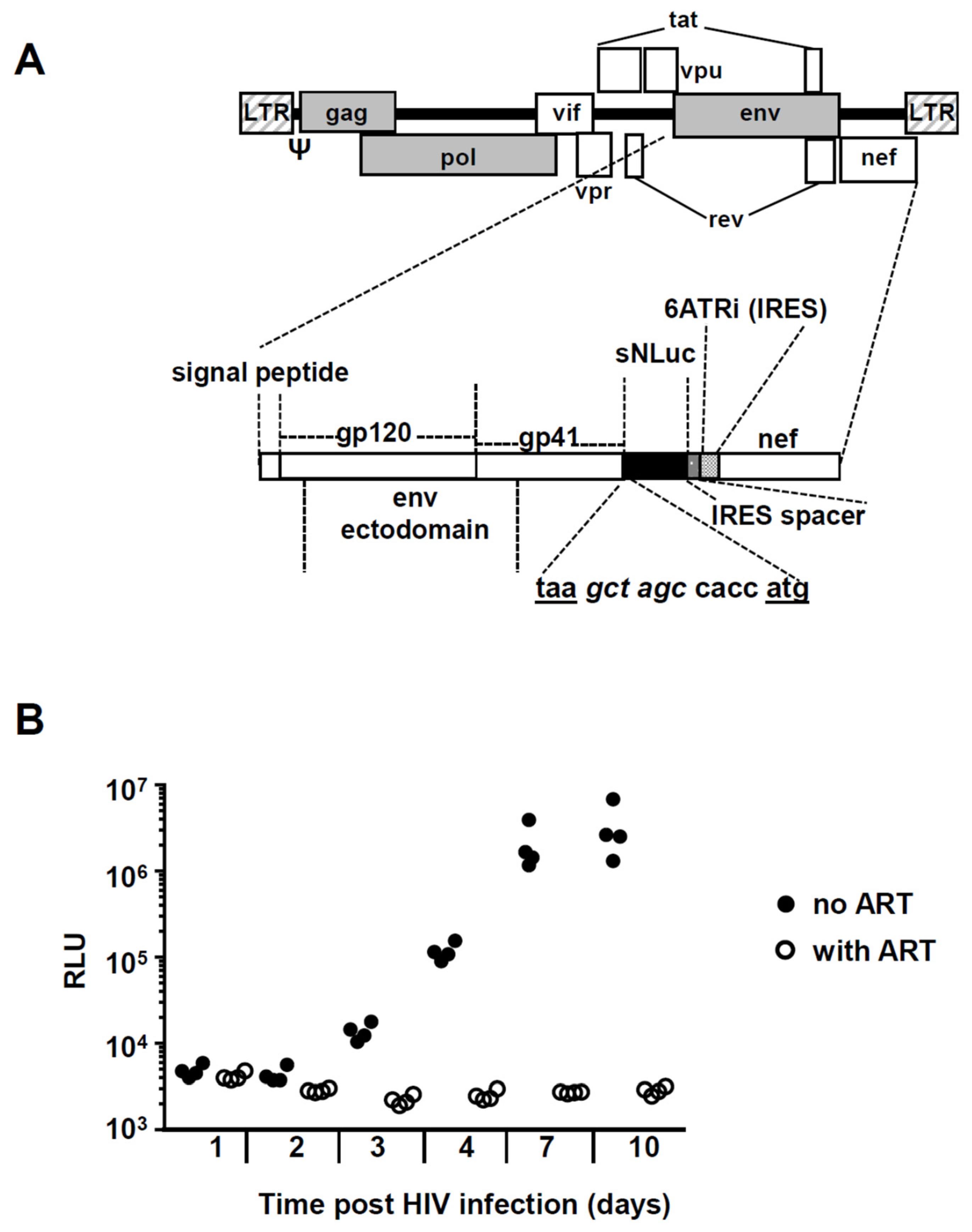
2.1. Generation of Replication Competent Nanoluciferase (NanoLuc®)-Expressing HIV-1 Reporter Virus
2.1.1. Description of Plasmid
2.1.2. Preparation of Viral Stock
2.2. Drugs
2.3. Cytokines
2.4. Cell Lines
2.4.1. TZM-bl Indicator Cells
2.4.2. J-Lat 11.1 Cells
2.4.3. J-Lat A7 Cells
2.4.4. U1 Cells
2.4.5. Isolation of Epithelial Cells and Leukocytes from the Female Genital Tract, and Monocytes, CD4+ T Cells and CD8+ T Cells from Peripheral Blood
2.4.6. Differentiation of Langerhans Cells from Hematopoietic Precursors
2.5. Western Blotting
2.6. In Vitro Infection of PBMC with HIV-1 Reporter Virus
2.7. Determination of Nanoluciferase Enzymatic Activity
2.8. TZM-bl Indicator Cell Infection
2.9. Quantification of Opioid Receptor Gene mRNA by RT-qPCR Assay
2.10. Statistical Analysis
3. Results
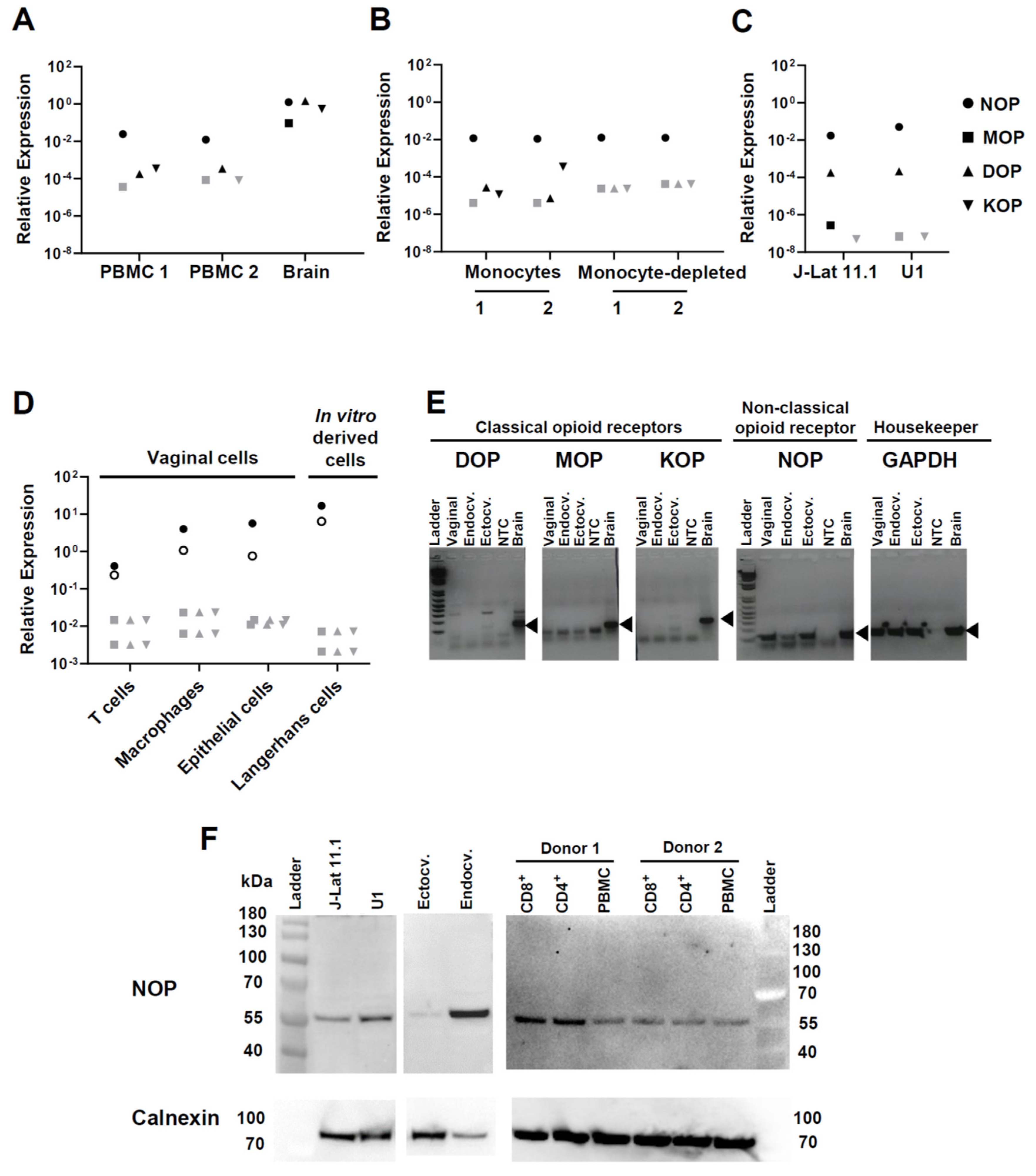
3.1. Opioid Receptor Gene Expression in Circulating and Mucosal Cell Types of Relevance for HIV Infection
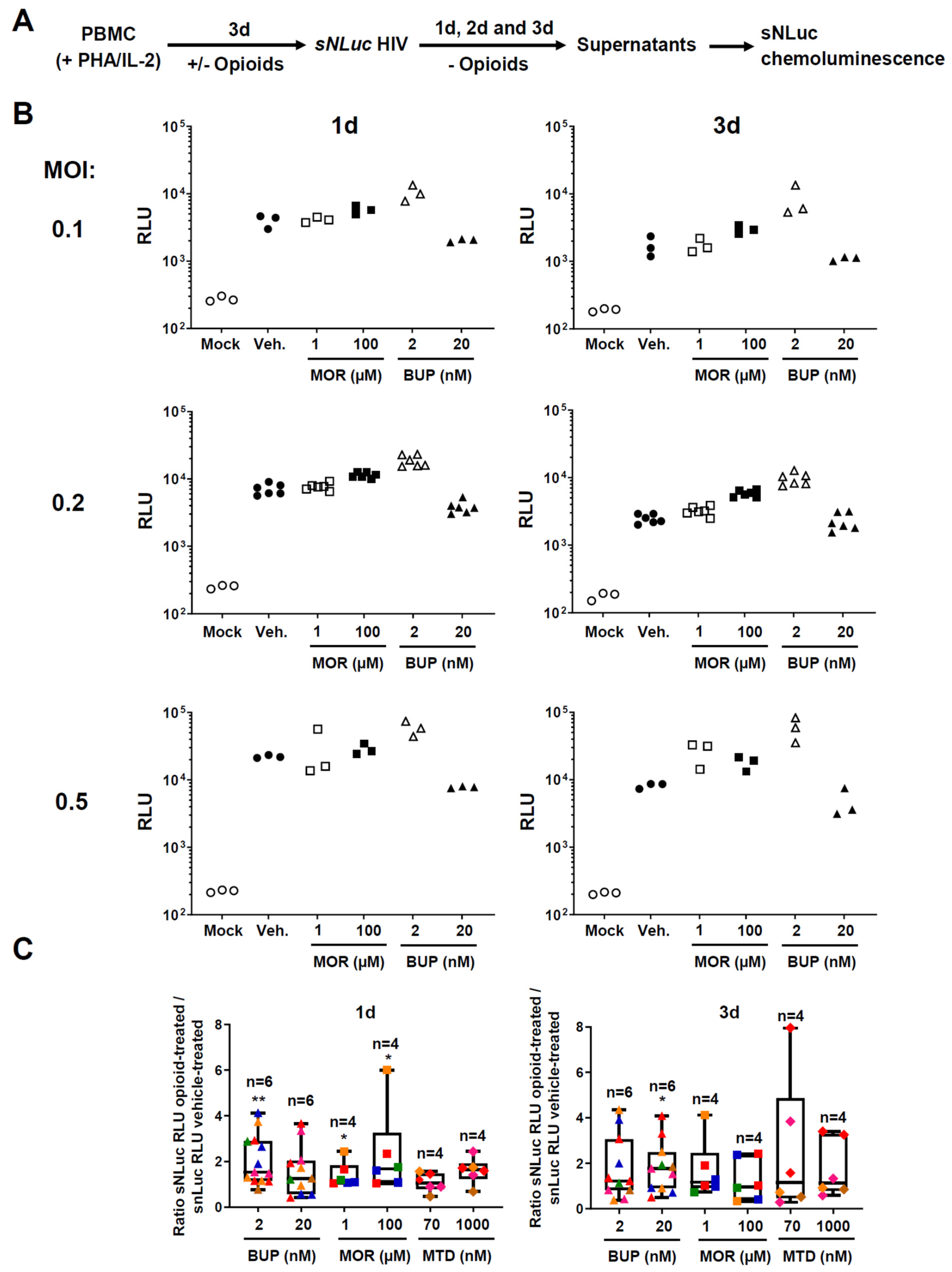
3.2. Effect of Buprenorphine, Methadone, and Morphine on HIV Replication in PBMC
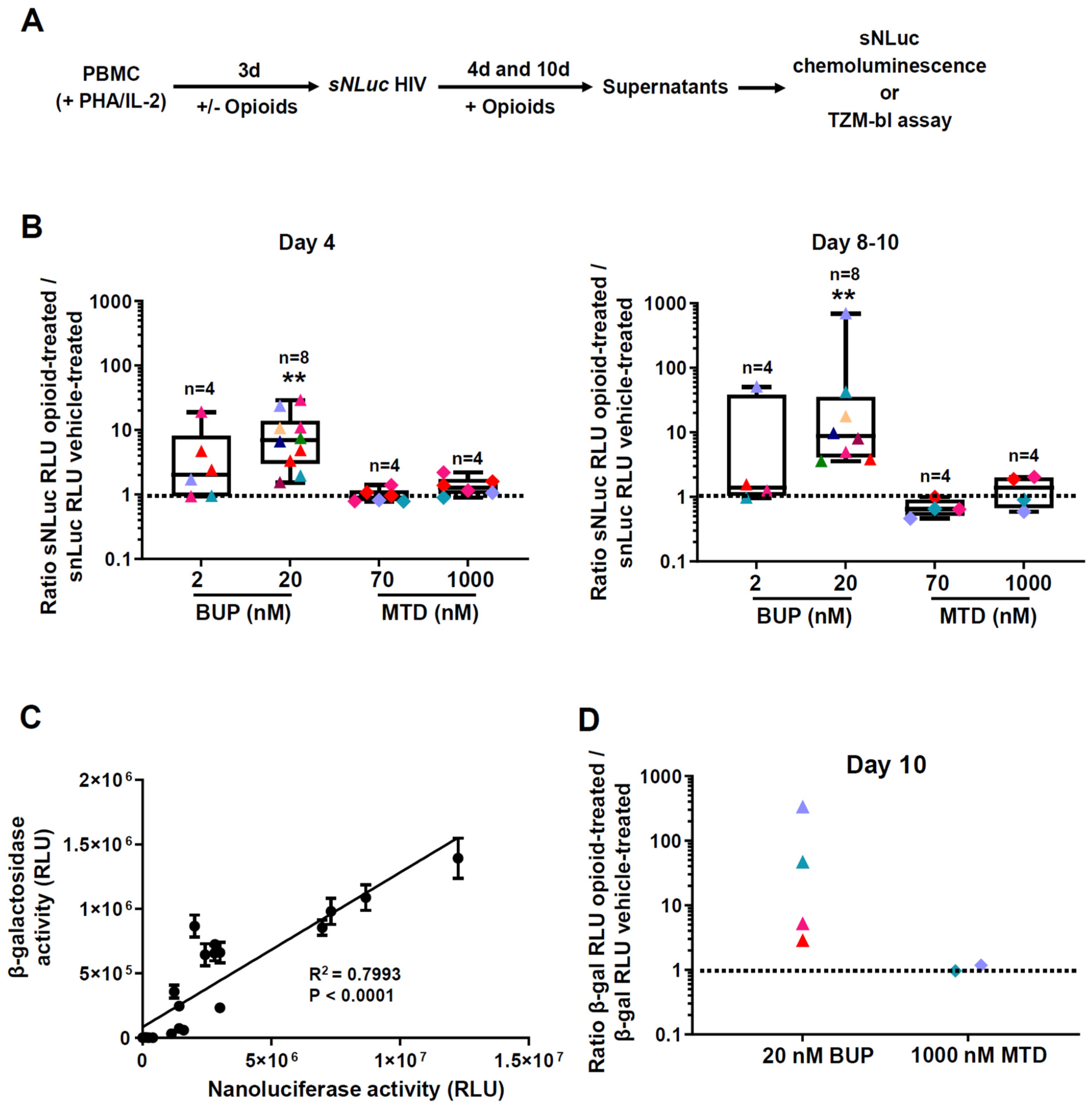
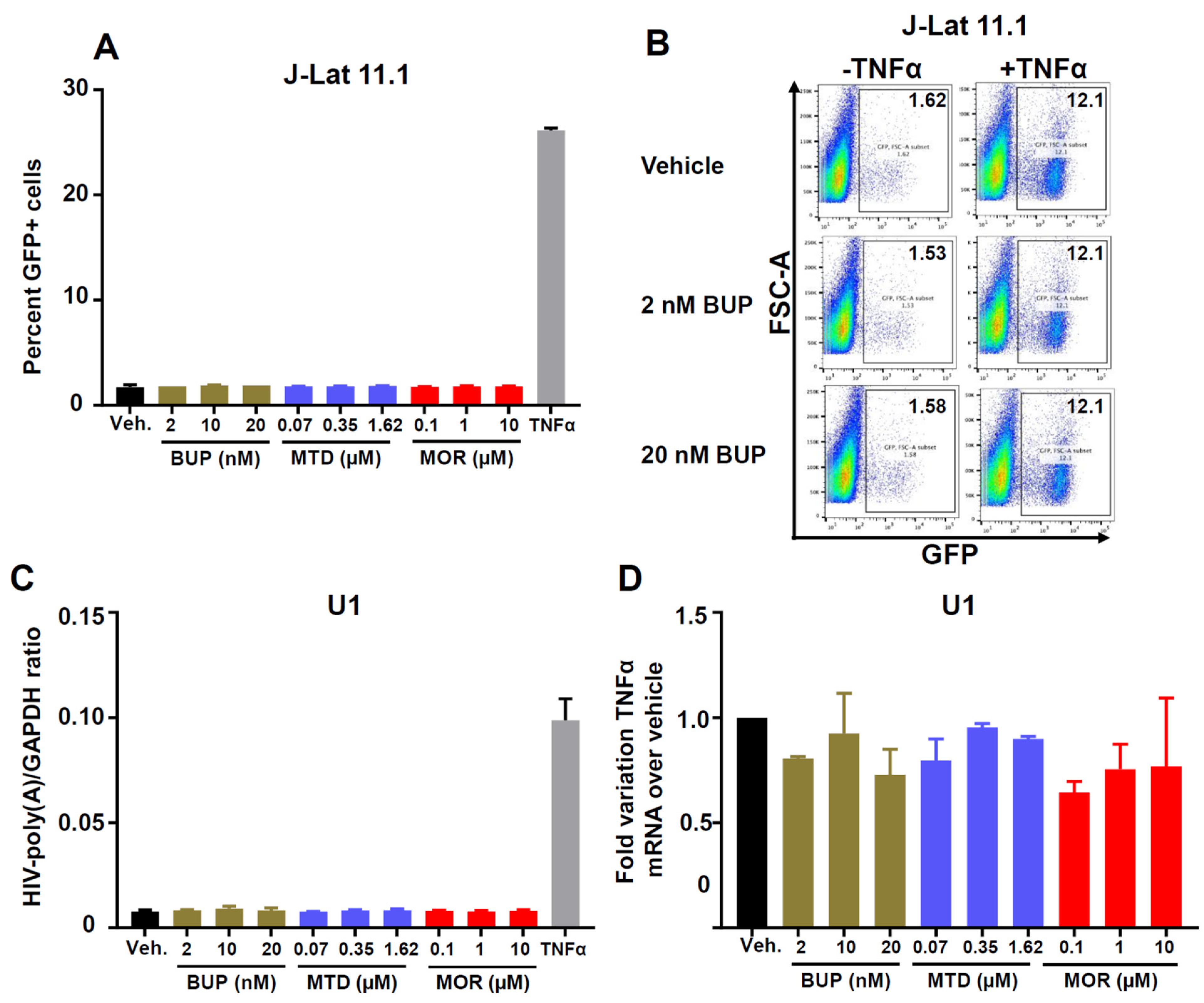
3.3. Effect of Methadone, Buprenorphine, and Morphine on HIV Reactivation from Latency
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Drug Report 2020. United Nations publication, Sales No. E.20.XI.6. 2020. Available online: https://wdr.unodc.org/wdr2020/index2020.html (accessed on 21 April 2021).
- Campbell, E.M.; Jia, H.; Shankar, A.; Hanson, D.; Luo, W.; Masciotra, S.; Owen, S.M.; Oster, A.M.; Galang, R.R.; Spiller, M.W.; et al. Detailed transmission network analysis of a large opiate-driven outbreak of HIV infection in the United States. J. Infect. Dis. 2017, 216, 1053–1062. [Google Scholar] [CrossRef]
- Kapadia, F.; Vlahov, D.; Donahoe, R.M.; Friedland, G. The role of substance abuse in HIV disease progression: Reconciling differences from laboratory and epidemiologic investigations. Clin. Infect. Dis. 2005, 41, 1027–1034. [Google Scholar] [CrossRef]
- Meng, J.; Sindberg, G.M.; Roy, S. Disruption of gut homeostasis by opioids accelerates HIV disease progression. Front. Microbiol. 2015, 6, 643. [Google Scholar] [CrossRef] [Green Version]
- Degenhardt, L.; Peacock, A.; Colledge, S.; Leung, J.; Grebely, J.; Vickerman, P.; Stone, J.; Cunningham, E.B.; Trickey, A.; Dumchev, K.; et al. Global prevalence of injecting drug use and sociodemographic characteristics and prevalence of HIV, HBV, and HCV in people who inject drugs: A multistage systematic review. Lancet Glob. Health 2017, 5, e1192–e1207. [Google Scholar] [CrossRef] [Green Version]
- Tun, W.; Sheehy, M.; Broz, D.; Okal, J.; Muraguri, N.; Raymond, H.F.; Musyoki, H.; Kim, A.A.; Muthui, M.; Geibel, S. HIV and STI prevalence and injection behaviors among people who inject drugs in Nairobi: Results from a 2011 bio-behavioral study using respondent-driven sampling. AIDS Behav. 2015, 19 (Suppl. 1), S24–S35. [Google Scholar] [CrossRef] [Green Version]
- Steele, A.D.; Henderson, E.E.; Rogers, T.J. Mu-opioid modulation of HIV-1 coreceptor expression and HIV-1 replication. Virology 2003, 309, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.J.; Li, Y.; Tian, S.; Wang, X.; Douglas, S.D.; Ho, W.Z. Morphine enhances HIV infection of human blood mononuclear phagocytes through modulation of beta-chemokines and CCR5 receptor. J. Investig. Med. 2002, 50, 435–442. [Google Scholar] [CrossRef]
- Li, Y.; Merrill, J.D.; Mooney, K.; Song, L.; Wang, X.; Guo, C.J.; Savani, R.C.; Metzger, D.S.; Douglas, S.D.; Ho, W.Z. Morphine enhances HIV infection of neonatal macrophages. Pediatr. Res. 2003, 54, 282–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, P.K.; Sharp, B.M.; Gekker, G.; Portoghese, P.S.; Sannerud, K.; Balfour, H.H., Jr. Morphine promotes the growth of HIV-1 in human peripheral blood mononuclear cell cocultures. AIDS 1990, 4, 869–873. [Google Scholar] [CrossRef]
- Schweitzer, C.; Keller, F.; Schmitt, M.P.; Jaeck, D.; Adloff, M.; Schmitt, C.; Royer, C.; Kirn, A.; Aubertin, A.M. Morphine stimulates HIV replication in primary cultures of human Kupffer cells. Res. Virol. 1991, 142, 189–195. [Google Scholar] [CrossRef]
- Suzuki, S.; Chuang, A.J.; Chuang, L.F.; Doi, R.H.; Chuang, R.Y. Morphine promotes simian acquired immunodeficiency syndrome virus replication in monkey peripheral mononuclear cells: Induction of CC chemokine receptor 5 expression for virus entry. J. Infect. Dis. 2002, 185, 1826–1829. [Google Scholar] [CrossRef] [PubMed]
- Prottengeier, J.; Koutsilieri, E.; Scheller, C. The effects of opioids on HIV reactivation in latently-infected T-lymphoblasts. AIDS Res. 2014, 11, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, P.K.; Gekker, G.; Hu, S.; Anderson, W.R.; Kravitz, F.; Portoghese, P.S.; Balfour, H.H., Jr.; Chao, C.C. Morphine amplifies HIV-1 expression in chronically infected promonocytes cocultured with human brain cells. J. Neuroimmunol. 1994, 50, 167–175. [Google Scholar] [CrossRef]
- Chao, C.C.; Gekker, G.; Sheng, W.S.; Hu, S.; Tsang, M.; Peterson, P.K. Priming effect of morphine on the production of tumor necrosis factor-alpha by microglia: Implications in respiratory burst activity and human immunodeficiency virus-1 expression. J. Pharm. Exp. 1994, 269, 198–203. [Google Scholar]
- Wang, X.; Ye, L.; Zhou, Y.; Liu, M.Q.; Zhou, D.J.; Ho, W.Z. Inhibition of anti-HIV microRNA expression: A mechanism for opioid-mediated enhancement of HIV infection of monocytes. Am. J. Pathol. 2011, 178, 41–47. [Google Scholar] [CrossRef]
- Szabo, I.; Wetzel, M.A.; Zhang, N.; Steele, A.D.; Kaminsky, D.E.; Chen, C.; Liu-Chen, L.Y.; Bednar, F.; Henderson, E.E.; Howard, O.M.; et al. Selective inactivation of CCR5 and decreased infectivity of R5 HIV-1 strains mediated by opioid-induced heterologous desensitization. J. Leukoc. Biol. 2003, 74, 1074–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaminsky, D.E.; Rogers, T.J. Nociceptin/orphanin FQ receptor-driven heterologous desensitization of the major HIV-1 co-receptor CXCR4. J. Neuroimmune Pharmacol. Off. J. Soc. NeuroImmune Pharmacol. 2011, 6, 546–550. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Torres, C.; Yamamura, Y.; Rodriguez, I.; Martinez, M.; Staprans, S.; Donahoe, R.M.; Kraiselburd, E.; Stephens, E.B.; Kumar, A. Modulation by morphine of viral set point in rhesus macaques infected with simian immunodeficiency virus and simian-human immunodeficiency virus. J. Virol. 2004, 78, 11425–11428. [Google Scholar] [CrossRef] [Green Version]
- Donahoe, R.M.; Vlahov, D. Opiates as potential cofactors in progression of HIV-1 infections to AIDS. J. Neuroimmunol. 1998, 83, 77–87. [Google Scholar] [CrossRef]
- Donahoe, R.M.; O’Neil, S.P.; Marsteller, F.A.; Novembre, F.J.; Anderson, D.C.; Lankford-Turner, P.; McClure, H.H. Probable deceleration of progression of Simian AIDS affected by opiate dependency: Studies with a rhesus macaque/SIVsmm9 model. J. Acquir. Immune Defic. Syndr. 2009, 50, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Tahamtan, A.; Tavakoli-Yaraki, M.; Mokhtari-Azad, T.; Teymoori-Rad, M.; Bont, L.; Shokri, F.; Salimi, V. Opioids and Viral Infections: A Double-Edged Sword. Front. Microbiol. 2016, 7, 970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Hashimi, M.; Scott, S.W.; Thompson, J.P.; Lambert, D.G. Opioids and immune modulation: More questions than answers. Br. J. Anaesth. 2013, 111, 80–88. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Liu, R.; Chen, C.; Ji, F.; Li, T. Opioid System Modulates the Immune Function: A Review. Transl. Perioper. Pain Med. 2016, 1, 5–13. [Google Scholar] [PubMed]
- Odunayo, A.; Dodam, J.R.; Kerl, M.E.; DeClue, A.E. Immunomodulatory effects of opioids. J. Vet. Emerg. Crit. Care 2010, 20, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Sacerdote, P. Opioid-induced immunosuppression. Curr. Opin. Support. Palliat. Care 2008, 2, 14–18. [Google Scholar] [CrossRef]
- Boland, J.W.; Foulds, G.A.; Ahmedzai, S.H.; Pockley, A.G. A preliminary evaluation of the effects of opioids on innate and adaptive human in vitro immune function. BMJ Support. Palliat. Care 2014, 4, 357–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Börner, C.; Lanciotti, S.; Koch, T.; Höllt, V.; Kraus, J. μ opioid receptor agonist-selective regulation of interleukin-4 in T lymphocytes. J. Neuroimmunol. 2013, 263, 35–42. [Google Scholar] [CrossRef]
- Cornwell, W.D.; Lewis, M.G.; Fan, X.; Rappaport, J.; Rogers, T.J. Effect of chronic morphine administration on circulating T cell population dynamics in rhesus macaques. J. Neuroimmunol. 2013, 265, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Bhargava, H.N.; Thomas, P.T.; Thorat, S.; House, R.V. Effects of morphine tolerance and abstinence on cellular immune function. Brain Res. 1994, 642, 1–10. [Google Scholar] [CrossRef]
- Yokota, T.; Uehara, K.; Nomoto, Y. Addition of noradrenaline to intrathecal morphine augments the postoperative suppression of natural killer cell activity. J. Anesth. 2004, 18, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Flores, R.; Suo, J.L.; Weber, R.J. Suppression of splenic macrophage functions following acute morphine action in the rat mesencephalon periaqueductal gray. Brain Behav. Immun. 1999, 13, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Barke, R.A.; Charboneau, R.; Roy, S. Morphine impairs host innate immune response and increases susceptibility to Streptococcus pneumoniae lung infection. J. Immunol. 2005, 174, 426–434. [Google Scholar] [CrossRef] [Green Version]
- Sacerdote, P.; Manfredi, B.; Mantegazza, P.; Panerai, A.E. Antinociceptive and immunosuppressive effects of opiate drugs: A structure-related activity study. Br. J. Pharm. 1997, 121, 834–840. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Shorter, D.; Kosten, T.R. Buprenorphine in the treatment of opioid addiction: Opportunities, challenges and strategies. Expert Opin. Pharm. 2014, 15, 2263–2275. [Google Scholar] [CrossRef]
- Neri, S.; Bruno, C.M.; Pulvirenti, D.; Malaguarnera, M.; Italiano, C.; Mauceri, B.; Abate, G.; Cilio, D.; Calvagno, S.; Tsami, A.; et al. Randomized clinical trial to compare the effects of methadone and buprenorphine on the immune system in drug abusers. Psychopharmacology 2005, 179, 700–704. [Google Scholar] [CrossRef]
- Riss, G.L.; Chang, D.I.; Wevers, C.; Westendorf, A.M.; Buer, J.; Scherbaum, N.; Hansen, W. Opioid maintenance therapy restores CD4+ T cell function by normalizing CD4+CD25(high) regulatory T cell frequencies in heroin user. Brain Behav. Immun. 2012, 26, 972–978. [Google Scholar] [CrossRef]
- LeVier, D.G.; Brown, R.D.; Musgrove, D.L.; Butterworth, L.F.; McCay, J.A.; White, K.L., Jr.; Fuchs, B.A.; Harris, L.S.; Munson, A.E. The effect of methadone on the immune status of B6C3F1 mice. Fundam. Appl. Toxicol. Off. J. Soc. Toxicol. 1995, 24, 275–284. [Google Scholar]
- Peterson, P.K.; Gekker, G.; Brummitt, C.; Pentel, P.; Bullock, M.; Simpson, M.; Hitt, J.; Sharp, B. Suppression of human peripheral blood mononuclear cell function by methadone and morphine. J. Infect. Dis. 1989, 159, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Chuang, L.F.; Lam, K.M.; Kung, H.F.; Wang, J.M.; Osburn, B.I.; Chuang, R.Y. Inhibition of chemokine-induced chemotaxis of monkey leukocytes by mu-opioid receptor agonists. In Vivo 1999, 13, 389–396. [Google Scholar]
- Gomez-Flores, R.; Weber, R.J. Differential effects of buprenorphine and morphine on immune and neuroendocrine functions following acute administration in the rat mesencephalon periaqueductal gray. Immunopharmacology 2000, 48, 145–156. [Google Scholar] [CrossRef]
- Walker, J.S.; Chandler, A.K.; Wilson, J.L.; Binder, W.; Day, R.O. Effect of mu-opioids morphine and buprenorphine on the development of adjuvant arthritis in rats. Inflamm. Res. 1996, 45, 557–563. [Google Scholar] [CrossRef]
- Martucci, C.; Panerai, A.E.; Sacerdote, P. Chronic fentanyl or buprenorphine infusion in the mouse: Similar analgesic profile but different effects on immune responses. Pain 2004, 110, 385–392. [Google Scholar] [CrossRef]
- Franchi, S.; Panerai, A.E.; Sacerdote, P. Buprenorphine ameliorates the effect of surgery on hypothalamus-pituitary-adrenal axis, natural killer cell activity and metastatic colonization in rats in comparison with morphine or fentanyl treatment. Brain Behav. Immun. 2007, 21, 767–774. [Google Scholar] [CrossRef]
- D’Elia, M.; Patenaude, J.; Hamelin, C.; Garrel, D.R.; Bernier, J. No detrimental effect from chronic exposure to buprenorphine on corticosteroid-binding globulin and corticosensitive immune parameters. Clin. Immunol. 2003, 109, 179–187. [Google Scholar] [CrossRef]
- Carrigan, K.A.; Saurer, T.B.; Ijames, S.G.; Lysle, D.T. Buprenorphine produces naltrexone reversible alterations of immune status. Int. Immunopharmacol. 2004, 4, 419–428. [Google Scholar] [CrossRef]
- Piersma, F.E.; Daemen, M.A.; Bogaard, A.E.; Buurman, W.A. Interference of pain control employing opioids in in vivo immunological experiments. Lab. Anim. 1999, 33, 328–333. [Google Scholar] [CrossRef]
- Felix, N.M.; Leal, R.O.; Goy-Thollot, I.; Walton, R.S.; Gil, S.A.; Mateus, L.M.; Matos, A.S.; Niza, M. Effects of buprenorphine in the adrenal, thyroid, and cytokine intra-operative responses in a rat model (Rattus norvegicus): A preliminary study. Iran. J. Basic Med. Sci. 2017, 20, 368–379. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X.; Tian, S.; Guo, C.J.; Douglas, S.D.; Ho, W.Z. Methadone enhances human immunodeficiency virus infection of human immune cells. J. Infect. Dis. 2002, 185, 118–122. [Google Scholar] [CrossRef] [Green Version]
- Astronomo, R.D.; Santra, S.; Ballweber-Fleming, L.; Westerberg, K.G.; Mach, L.; Hensley-McBain, T.; Sutherland, L.; Mildenberg, B.; Morton, G.; Yates, N.L.; et al. Neutralization Takes Precedence Over IgG or IgA Isotype-related Functions in Mucosal HIV-1 Antibody-mediated Protection. EBioMedicine 2016, 14, 97–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberti, M.O.; Jones, J.J.; Miglietta, R.; Ding, H.; Bakshi, R.K.; Edmonds, T.G.; Kappes, J.C.; Ochsenbauer, C. Optimized Replicating Renilla Luciferase Reporter HIV-1 Utilizing Novel Internal Ribosome Entry Site Elements for Native Nef Expression and Function. AIDS Res. Hum. Retrovir. 2015, 31, 1278–1296. [Google Scholar] [CrossRef] [Green Version]
- Cavrois, M.; Banerjee, T.; Mukherjee, G.; Raman, N.; Hussien, R.; Rodriguez, B.A.; Vasquez, J.; Spitzer, M.H.; Lazarus, N.H.; Jones, J.J.; et al. Mass Cytometric Analysis of HIV Entry, Replication, and Remodeling in Tissue CD4+ T Cells. Cell Rep. 2017, 20, 984–998. [Google Scholar] [CrossRef] [Green Version]
- Prévost, J.; Richard, J.; Medjahed, H.; Alexander, A.; Jones, J.; Kappes, J.C.; Ochsenbauer, C.; Finzi, A. Incomplete Downregulation of CD4 Expression Affects HIV-1 Env Conformation and Antibody-Dependent Cellular Cytotoxicity Responses. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Ventura, J.D.; Beloor, J.; Allen, E.; Zhang, T.; Haugh, K.A.; Uchil, P.D.; Ochsenbauer, C.; Kieffer, C.; Kumar, P.; Hope, T.J.; et al. Longitudinal bioluminescent imaging of HIV-1 infection during antiretroviral therapy and treatment interruption in humanized mice. PLoS Pathog. 2019, 15, e1008161. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Decker, J.M.; Liu, H.; Zhang, Z.; Arani, R.B.; Kilby, J.M.; Saag, M.S.; Wu, X.; Shaw, G.M.; Kappes, J.C. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 2002, 46, 1896–1905. [Google Scholar] [CrossRef] [Green Version]
- Jordan, A.; Bisgrove, D.; Verdin, E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003, 22, 1868–1877. [Google Scholar] [CrossRef] [Green Version]
- Folks, T.M.; Justement, J.; Kinter, A.; Dinarello, C.A.; Fauci, A.S. Cytokine-induced expression of HIV-1 in a chronically infected promonocyte cell line. Science 1987, 238, 800–802. [Google Scholar] [CrossRef] [PubMed]
- McElrath, M.J.; Ballweber, L.; Terker, A.; Kreger, A.; Sakchalathorn, P.; Robinson, B.; Fialkow, M.; Lentz, G.; Hladik, F. Ex vivo comparison of microbicide efficacies for preventing HIV-1 genomic integration in intraepithelial vaginal cells. Antimicrob. Agents Chemother. 2010, 54, 763–772. [Google Scholar] [CrossRef] [Green Version]
- Hladik, F.; Burgener, A.; Ballweber, L.; Gottardo, R.; Vojtech, L.; Fourati, S.; Dai, J.Y.; Cameron, M.J.; Strobl, J.; Hughes, S.M.; et al. Mucosal effects of tenofovir 1% gel. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Gornalusse, G.G.; Valdez, R.; Fenkart, G.; Vojtech, L.; Fleming, L.M.; Pandey, U.; Hughes, S.M.; Levy, C.N.; Dela Cruz, E.J.; Calienes, F.L.; et al. Mechanisms of Endogenous HIV-1 Reactivation by Endocervical Epithelial Cells. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- McKinnon, L.R.; Hughes, S.M.; De Rosa, S.C.; Martinson, J.A.; Plants, J.; Brady, K.E.; Gumbi, P.P.; Adams, D.J.; Vojtech, L.; Galloway, C.G.; et al. Optimizing viable leukocyte sampling from the female genital tract for clinical trials: An international multi-site study. PLoS ONE 2014, 9, e85675. [Google Scholar] [CrossRef] [PubMed]
- Gatti, E.; Velleca, M.a.; Biedermann, B.C.; Ma, W.; Unternaehrer, J.; Ebersold, M.W.; Medzhitov, R.; Pober, J.S.; Mellman, I. Large-scale culture and selective maturation of human Langerhans cells from granulocyte colony-stimulating factor-mobilized CD34+ progenitors. J. Immunol. 2000, 164, 3600–3607. [Google Scholar] [CrossRef] [Green Version]
- Mann, A.; Mouledous, L.; Froment, C.; O’Neill, P.R.; Dasgupta, P.; Gunther, T.; Brunori, G.; Kieffer, B.L.; Toll, L.; Bruchas, M.R.; et al. Agonist-selective NOP receptor phosphorylation correlates in vitro and in vivo and reveals differential post-activation signaling by chemically diverse agonists. Sci. Signal. 2019, 12, eaau8072. [Google Scholar] [CrossRef]
- Connery, H.S. Medication-assisted treatment of opioid use disorder: Review of the evidence and future directions. Harv. Rev. Psychiatry 2015, 23, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.P.; Thompson, J.P.; McDonald, J.; Barnes, T.A.; Cote, T.; Rowbotham, D.J.; Lambert, D.G. Human peripheral blood mononuclear cells express nociceptin/orphanin FQ, but not mu, delta, or kappa opioid receptors. Anesth. Analg. 2007, 105, 998–1005. [Google Scholar] [CrossRef]
- Al-Hashimi, M.; McDonald, J.; Thompson, J.P.; Lambert, D.G. Evidence for nociceptin/orphanin FQ (NOP) but not micro (MOP), delta (DOP) or kappa (KOP) opioid receptor mRNA in whole human blood. Br. J. Anaesth. 2016, 116, 423–429. [Google Scholar] [CrossRef] [Green Version]
- Peluso, J.; LaForge, K.S.; Matthes, H.W.; Kreek, M.J.; Kieffer, B.L.; Gaveriaux-Ruff, C. Distribution of nociceptin/orphanin FQ receptor transcript in human central nervous system and immune cells. J. Neuroimmunol. 1998, 81, 184–192. [Google Scholar] [CrossRef]
- Wick, M.J.; Minnerath, S.R.; Roy, S.; Ramakrishnan, S.; Loh, H.H. Expression of alternate forms of brain opioid ‘orphan’ receptor mRNA in activated human peripheral blood lymphocytes and lymphocytic cell lines. Brain Res. Mol. Brain Res. 1995, 32, 342–347. [Google Scholar] [CrossRef]
- Kaminsky, D.E.; Rogers, T.J. Suppression of CCL2/MCP-1 and CCL5/RANTES expression by nociceptin in human monocytes. J. Neuroimmune Pharmacol. 2008, 3, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Arjomand, J.; Cole, S.; Evans, C.J. Novel orphanin FQ/nociceptin transcripts are expressed in human immune cells. J. Neuroimmunol. 2002, 130, 100–108. [Google Scholar] [CrossRef]
- Machelska, H.; Celik, M.O. Opioid Receptors in Immune and Glial Cells-Implications for Pain Control. Front. Immunol. 2020, 11, 300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, M.; Mirmohammadsadegh, A.; Franz, B.; Blanke, J.; Hengge, U.R. Opioid receptors on white blood cells: Effect of HIV infection and methadone treatment. Pain 2002, 98, 187–194. [Google Scholar] [CrossRef]
- Sharp, B.M.; McAllen, K.; Gekker, G.; Shahabi, N.A.; Peterson, P.K. Immunofluorescence detection of delta opioid receptors (DOR) on human peripheral blood CD4+ T cells and DOR-dependent suppression of HIV-1 expression. J. Immunol. 2001, 167, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.C.; Gekker, G.; Sheng, W.S.; Hu, S.; Peterson, P.K. U50488 inhibits HIV-1 expression in acutely infected monocyte-derived macrophages. Drug Alcohol Depend. 2001, 62, 149–154. [Google Scholar] [CrossRef]
- Chao, C.C.; Gekker, G.; Hu, S.; Sheng, W.S.; Shark, K.B.; Bu, D.F.; Archer, S.; Bidlack, J.M.; Peterson, P.K. kappa opioid receptors in human microglia downregulate human immunodeficiency virus 1 expression. Proc. Natl. Acad. Sci. USA 1996, 93, 8051–8056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Human Protein Atlas. Available online: www.proteinatlas.org (accessed on 21 April 2021).
- Hruz, T.; Laule, O.; Szabo, G.; Wessendorp, F.; Bleuler, S.; Oertle, L.; Widmayer, P.; Gruissem, W.; Zimmermann, P. Genevestigator v3: A reference expression database for the meta-analysis of transcriptomes. Adv. Bioinform. 2008, 2008, 420747. [Google Scholar] [CrossRef] [PubMed]
- Bird, M.F.; Guerrini, R.; Willets, J.M.; Thompson, J.P.; Calo, G.; Lambert, D.G. Nociceptin/Orphanin FQ (N/OFQ) conjugated to ATTO594: A novel fluorescent probe for the N/OFQ (NOP) receptor. Br. J. Pharm. 2018, 175, 4496–4506. [Google Scholar] [CrossRef]
- Dever, S.M.; Xu, R.; Fitting, S.; Knapp, P.E.; Hauser, K.F. Differential expression and HIV-1 regulation of mu-opioid receptor splice variants across human central nervous system cell types. J. Neurovirol. 2012, 18, 181–190. [Google Scholar] [CrossRef]
- Regan, P.M.; Langford, D.; Khalili, K. Regulation and Functional Implications of Opioid Receptor Splicing in Opioid Pharmacology and HIV Pathogenesis. J. Cell. Physiol. 2016, 231, 976–985. [Google Scholar] [CrossRef] [Green Version]
- Klukovits, A.; Tekes, K.; Gunduz Cinar, O.; Benyhe, S.; Borsodi, A.; Deak, B.H.; Hajagos-Toth, J.; Verli, J.; Falkay, G.; Gaspar, R. Nociceptin inhibits uterine contractions in term-pregnant rats by signaling through multiple pathways. Biol. Reprod. 2010, 83, 36–41. [Google Scholar] [CrossRef] [Green Version]
- Deak, B.H.; Klukovits, A.; Tekes, K.; Ducza, E.; Falkay, G.; Gaspar, R. Nocistatin inhibits pregnant rat uterine contractions in vitro: Roles of calcitonin gene-related peptide and calcium-dependent potassium channel. Eur. J. Pharm. 2013, 714, 96–104. [Google Scholar] [CrossRef] [Green Version]
- El-Hage, N.; Wu, G.; Wang, J.; Ambati, J.; Knapp, P.E.; Reed, J.L.; Bruce-Keller, A.J.; Hauser, K.F. HIV-1 Tat and opiate-induced changes in astrocytes promote chemotaxis of microglia through the expression of MCP-1 and alternative chemokines. Glia 2006, 53, 132–146. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Tan, N.; Douglas, S.D.; Zhang, T.; Wang, Y.J.; Ho, W.Z. Morphine inhibits CD8+ T cell-mediated, noncytolytic, anti-HIV activity in latently infected immune cells. J. Leukoc. Biol. 2005, 78, 772–776. [Google Scholar] [CrossRef] [Green Version]
- Squinto, S.P.; Mondal, D.; Block, A.L.; Prakash, O. Morphine-induced transactivation of HIV-1 LTR in human neuroblastoma cells. AIDS Res. Hum. Retrovir. 1990, 6, 1163–1168. [Google Scholar] [CrossRef]
- Skopp, G.; Ganssmann, B.; Cone, E.J.; Aderjan, R. Plasma concentrations of heroin and morphine-related metabolites after intranasal and intramuscular administration. J. Anal. Toxicol. 1997, 21, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Park, H.S.; Kim, J.H.; Kim, Y.J.; Kim, D.Y. Plasma Concentrations of Morphine during Postoperative Pain Control. Korean J. Pain 2011, 24, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Kidd, S.; Brennan, S.; Stephen, R.; Minns, R.; Beattie, T. Comparison of morphine concentration-time profiles following intravenous and intranasal diamorphine in children. Arch. Dis. Child. 2009, 94, 974–978. [Google Scholar] [CrossRef] [PubMed]
- Jaureguiberry-Bravo, M.; Lopez, L.; Berman, J.W. Frontline Science: Buprenorphine decreases CCL2-mediated migration of CD14(+) CD16(+) monocytes. J. Leukoc. Biol. 2018, 104, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Carvallo, L.; Lopez, L.; Che, F.Y.; Lim, J.; Eugenin, E.A.; Williams, D.W.; Nieves, E.; Calderon, T.M.; Madrid-Aliste, C.; Fiser, A.; et al. Buprenorphine decreases the CCL2-mediated chemotactic response of monocytes. J. Immunol. 2015, 194, 3246–3258. [Google Scholar] [CrossRef] [Green Version]
- Toda, E.; Terashima, Y.; Sato, T.; Hirose, K.; Kanegasaki, S.; Matsushima, K. FROUNT is a common regulator of CCR2 and CCR5 signaling to control directional migration. J. Immunol. 2009, 183, 6387–6394. [Google Scholar] [CrossRef] [Green Version]
- Bloms-Funke, P.; Gillen, C.; Schuettler, A.J.; Wnendt, S. Agonistic effects of the opioid buprenorphine on the nociceptin/OFQ receptor. Peptides 2000, 21, 1141–1146. [Google Scholar] [CrossRef]
- Khanna, I.K.; Pillarisetti, S. Buprenorphine—An attractive opioid with underutilized potential in treatment of chronic pain. J. Pain Res. 2015, 8, 859–870. [Google Scholar] [CrossRef] [Green Version]
- Knapman, A.; Santiago, M.; Connor, M. Buprenorphine signalling is compromised at the N40D polymorphism of the human mu opioid receptor in vitro. Br. J. Pharm. 2014, 171, 4273–4288. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Young, B.; Pletcher, M.T.; Heilig, M.; Wahlestedt, C. Association between the nociceptin receptor gene (OPRL1) single nucleotide polymorphisms and alcohol dependence. Addict. Biol. 2008, 13, 88–94. [Google Scholar] [CrossRef]
- Kasai, S.; Nishizawa, D.; Hasegawa, J.; Sato, N.; Tanioka, F.; Sugimura, H.; Ikeda, K. Nociceptin/orphanin FQ receptor gene variation is associated with smoking status in Japanese. Pharmacogenomics 2016, 17, 1441–1451. [Google Scholar] [CrossRef]
- Inturrisi, C.E.; Verebely, K. The levels of methadone in the plasma in methadone maintenance. Clin. Pharm. 1972, 13, 633–637. [Google Scholar] [CrossRef]
- Bullingham, R.E.; McQuay, H.J.; Porter, E.J.; Allen, M.C.; Moore, R.A. Sublingual buprenorphine used postoperatively: Ten hour plasma drug concentration analysis. Br. J. Clin. Pharm. 1982, 13, 665–673. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Jones, J.G.; Robinson, P.; Henson, R.; Biggs, S.R.; Taylor, T. Plasma concentration and disposition of buprenorphine after intravenous and intramuscular doses to baboons. Eur. J. Drug Metab. Pharmacokinet. 1980, 5, 233–239. [Google Scholar] [CrossRef]
- Carrieri, M.P.; Vlahov, D.; Dellamonica, P.; Gallais, H.; Lepeu, G.; Spire, B.; Obadia, Y. Use of buprenorphine in HIV-infected injection drug users: Negligible impact on virologic response to HAART. The Manif-2000 Study Group. Drug Alcohol Depend. 2000, 60, 51–54. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gornalusse, G.G.; Vojtech, L.N.; Levy, C.N.; Hughes, S.M.; Kim, Y.; Valdez, R.; Pandey, U.; Ochsenbauer, C.; Astronomo, R.; McElrath, J.; et al. Buprenorphine Increases HIV-1 Infection In Vitro but Does Not Reactivate HIV-1 from Latency. Viruses 2021, 13, 1472. https://doi.org/10.3390/v13081472
Gornalusse GG, Vojtech LN, Levy CN, Hughes SM, Kim Y, Valdez R, Pandey U, Ochsenbauer C, Astronomo R, McElrath J, et al. Buprenorphine Increases HIV-1 Infection In Vitro but Does Not Reactivate HIV-1 from Latency. Viruses. 2021; 13(8):1472. https://doi.org/10.3390/v13081472
Chicago/Turabian StyleGornalusse, Germán Gustavo, Lucia N. Vojtech, Claire N. Levy, Sean M. Hughes, Yeseul Kim, Rogelio Valdez, Urvashi Pandey, Christina Ochsenbauer, Rena Astronomo, Julie McElrath, and et al. 2021. "Buprenorphine Increases HIV-1 Infection In Vitro but Does Not Reactivate HIV-1 from Latency" Viruses 13, no. 8: 1472. https://doi.org/10.3390/v13081472