1. Introduction
Chronic hepatitis following hepatitis B (HBV) and D (HDV) virus infections is the main cause of hepatocellular carcinoma (HCC) and liver cirrhosis. Even the availability of vaccination does not prevent the more than 800,000 deaths annually from the long-term effects of chronic liver inflammation associated with HBV/HDV infections [
1]. Both viruses are coated with identical envelope proteins that are coded by the 3.2 kb DNA genome of HBV [
2]. The RNA genome of HDV does not code for any envelope protein. Therefore, HDV acts as a so-called satellite virus and makes use of the envelope proteins derived from the HBV genome [
3,
4]. This is the reason why HDV can only spread in the presence of an HBV infection. In addition, the interaction of both viruses with their cellular entry receptor NTCP is based on the common envelope proteins (HBs) [
5]. NTCP (Na
+/taurocholate co-transporting polypeptide, gene symbol
SLC10A1) represents a physiologically important hepatic bile acid transporter and was also identified as the hepatic entry receptor for HBV and HDV. High affinity binding of both viruses to NTCP is mediated by the myristoylated preS1 domain (myr-preS1
2–48 lipopeptide), consisting of the 2–48 N-terminal amino acids of the large surface protein (LHBs). As a common but mostly non-curative therapy for virus-related chronic hepatitis, nucleos(t)ide-analogue (NUC) reverse transcriptase inhibitors (for HBV) and interferon (for both HBV/HDV) are used. Unfortunately, interferon therapy is highly prone to adverse drug reactions and NUCs have to be given life-long [
6,
7].
Identification of NTCP as an hepatic receptor for HBV/HDV in 2012 enabled the development of NTCP inhibitors as HBV/HDV entry inhibitors [
8,
9]. The mentioned myr-preS1
2–48 lipopeptide itself has the ability to block in vitro HBV/HDV infection with inhibitory constants (IC
50) in the low nanomolar range [
4]. Based on that, a synthetic analogue of this lipopeptide (Hepcludex
®) has been developed and was recently approved as first HDV entry inhibitor interacting with NTCP [
10]. Furthermore, numerous studies identified novel chemical entry inhibitors for HBV and HDV by screening for bile salt transport inhibitors [
11] or by screening for inhibitors of myr-preS1
2–48 lipopeptide attachment and/or in vitro HBV/HDV infection [
12] in appropriate hepatoma cell culture models overexpressing NTCP. However, to date, none of these small molecules have been able to enter the clinical development phase so far.
In previous studies, we identified individual compounds from two different classes (betulin and propanolamine derivatives) that were quite potent for myr-preS1
2–48 lipopeptide binding inhibition and significantly blocked in vitro HDV infection of NTCP-expressing HepG2 cells [
13,
14]. In the present study, we aimed to expand the group of small molecule NTCP inhibitors by pharmacophore-based virtual screening (VS) of compound libraries.
A well-established method of identifying novel inhibitor candidates for cellular drug targets is high-throughput screening (HTS) of chemical libraries. This technique can rapidly generate data of large subsets of molecules using automated experimental assays [
15]. However, hit rates of HTS are only between 0.01% and 0.1% [
16], which leads to immense drug discovery costs [
17]. An alternative is given by so-called quantitative structure–activity relationship (QSAR) analysis as a ligand-based method for drug design [
18]. In principle, this represents a bioinformatic method for building mathematical models that describe the correlation between physicochemical properties of ligands and continuous (IC
50, EC
50,
Ki, etc.) or categorical (active, inactive, toxic, nontoxic, etc.) properties, by using statistical regression techniques [
19]. Nowadays, these models are used for VS approaches to predicting activities for compounds of large chemical databases (e.g., ZINC
15 [
20]). Only substances with the best predicted activities are then selected for experimental validation [
21]. Hit rates of this method range from 1% to 40% depending on the predictability of the generated model [
22]. Compared to experimental HTS, QSAR-based VS of chemical libraries often results in higher hit rates of biologically active compounds at lower costs [
22]. In the present study, we demonstrate that by QSAR-based VS novel small molecule inhibitors of NTCP can be identified that indeed showed proof-of-concept inhibition of HDV infection.
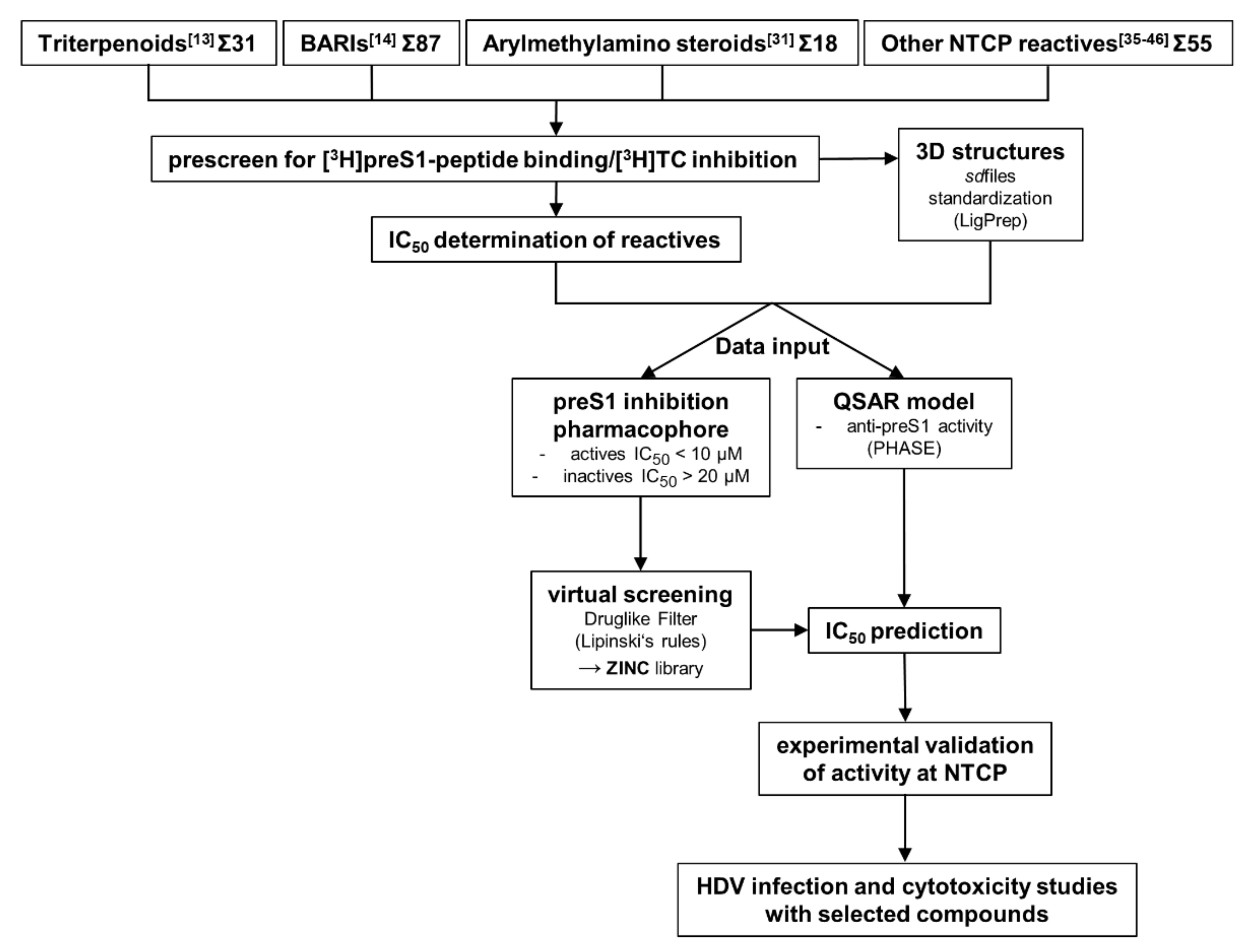
3. Results
The most important factor for the predictability of pharmacophore and QSAR models is the quality of data input [
34]. To ensure input of data from congeneric experiments, we performed all experiments with the identical cell line and protocol, as described before [
13,
14]. Namely, we used all test compounds as inhibitors for [
3H]preS1-peptide binding inhibition to NTCP and [
3H]TC transport inhibition via NTCP, both in NTCP-expressing HEK293 cells. These experiments were performed with four different groups of test compounds, including 31 betulin-derived triterpenoids [
13], and 87 propanolamine derivatives [
14]. Both groups of compounds were analyzed before as novel NTCP inhibitors in our laboratory. In addition, a set of 18 arylmethylamino steroids that previously showed antiparasitic activity against
Plasmodium falciparum and
Schistosoma mansoni [
31] was used for NTCP inhibition for the first time in the present study (
Supplementary Figures S1 and S2). And finally, a group of 55 structurally unrelated compounds that were reported as NTCP inhibitors in the literature [
35,
36,
37,
38,
39,
40,
41,
42,
43,
44,
45,
46] were included in the [
3H]preS1-peptide binding inhibition experiments.
Figure 1 gives an overview about the workflow of the present study.
In a first step, all compounds were used at 100 µM inhibitory concentration in binding experiments with 5 nM [
3H]preS1-peptide and transport experiments with 1 µM [
3H]TC in NTCP-overexpressing HEK293 cells (
Supplementary Figure S3). For all compounds that showed more than 50% inhibition of [
3H]preS1-peptide binding inhibition, detailed half-maximal inhibitory concentrations (IC
50) were determined. However, for some of the compounds a sigmoidal concentration-dependent dose-response relationship could not be determined experimentally. Therefore, these compounds were removed from the dataset. This was important to avoid distortion of subsequent models due to non-valid IC
50 data. In total, a data set consisting of 85 compounds with valid IC
50 data could be compiled. IC
50 values for the betulin [
13] and propanolamine [
14] derivatives were taken from previous studies of our laboratory. The inhibition pattern of the arylmethylamino steroids is presented in detail in
Supplementary Figure S1. The IC
50 values for [
3H]preS1-peptide binding inhibition of this compound class ranged from 8–251 µM. The most potent compound was steroid 12c with an IC
50 for [
3H]preS1-peptide binding inhibition of 8 µM. Structures of the three most potent [
3H]preS1-peptide binding inhibitors of this compound class are depicted in
Supplementary Figure S2.
For QSAR modeling, all IC
50 values were transformed by −log(IC
50 [M]) conversion into binding affinity values as indicated in
Table 1 In addition, data of the chemical structures were collected and were saved as 2D
sdfiles. For curation and standardization of the chemical structures, LigPrep [
32] was used (encoded in the software of the Schrödinger Suite which can be executed through the Maestro graphical user interface). In total, the IC
50 values for [
3H]preS1-peptide binding inhibition from the 85 data set compounds revealed binding affinities between 1 (IC
50 > 1000 µM) and5.699 (IC
50 = 2 µM). The compounds then were divided into a training set to generate the QSAR model (59 compounds,
Table 1) and a test set to validate the QSAR model (26 compounds, Table 4) as described in the Material and Methods
Section 2.8.
The atom-based QSAR model, illustrated in
Figure 2, was built using PHASE [
33]. This model describes three-dimensionally all necessary features to block [
3H]preS1-peptide binding to NTCP. Seven scenarios with different numbers of partial least squares (PLS) factors of the chosen regression model were generated and statistically analyzed (
Table 2). A number of four PLS factors revealed the highest value of Pearson-r for the predicted activities of the test set of 0.4614. Therefore, this model composition was chosen for all further investigations. The standard deviation (SD) of the regression for the chosen model was at 0.153 with a coefficient of determination (R
2) of 0.9519. The stability index of −0.242 of this model illustrates that the texture of the model is strongly dependent on the training set composition. The variance ratio (F) of 357.3 indicates statistically significant regression and the significance level (P) of −41 indicates a great degree of confidence for the variance ratio (
Table 2).
Distribution of the atom types of the QSAR model is shown in
Table 3 for all seven scenarios of PLS factors. Listed are the percentages of H-bond donor, hydrophobic/non-polar, negative/positive ionic, electron withdrawing, and other regions in the model. The proportions of these attributes point to the relative importance of each attribute for the NTCP inhibitory potency of the respective compound. Interestingly, the attribute proportions did not strongly differ between the respective numbers of PLS factors and were calculated to ~5% H-bond donor, ~60% hydrophobic/non-polar, <1% negative or positive ionic, and ~30% electron withdrawing (
Table 3). This means that the amount and distribution of hydrophobic or nonpolar regions is the most important factor for the potency of the inhibitor, while positive or negative ionic residues are of low importance. As expected, there was a strong correlation between the experimentally measured and the QSAR-based predicted binding affinity of the 59 training set compounds with R
2 of 0.9591 and slope of 0.959 (
Figure 3A). Based on this, the binding affinities of the 26 test-set compounds were predicted via the atom-based QSAR model and these ranged from 0.918 (IC
50 > 1000 µM) to 5.836 (IC
50 = 1.5 µM) with a mean error of prediction of −0.005 (
Table 4).
Figure 3B shows the correlation between the experimentally measured and the QSAR-based predicted binding affinities of the 26 test-set compounds that revealed R
2 of 0.2163 and slope of 0.2439. For five out of the 26 compounds the prediction was quite exact, representing ~20% high-level predictability These compounds are steroid 7s (error −0.01), raloxifene (error 0.057), compound S985852 (error −0.08), steroid 2c (error 0.102), and pioglitazone (error 0.119) (
Table 4).
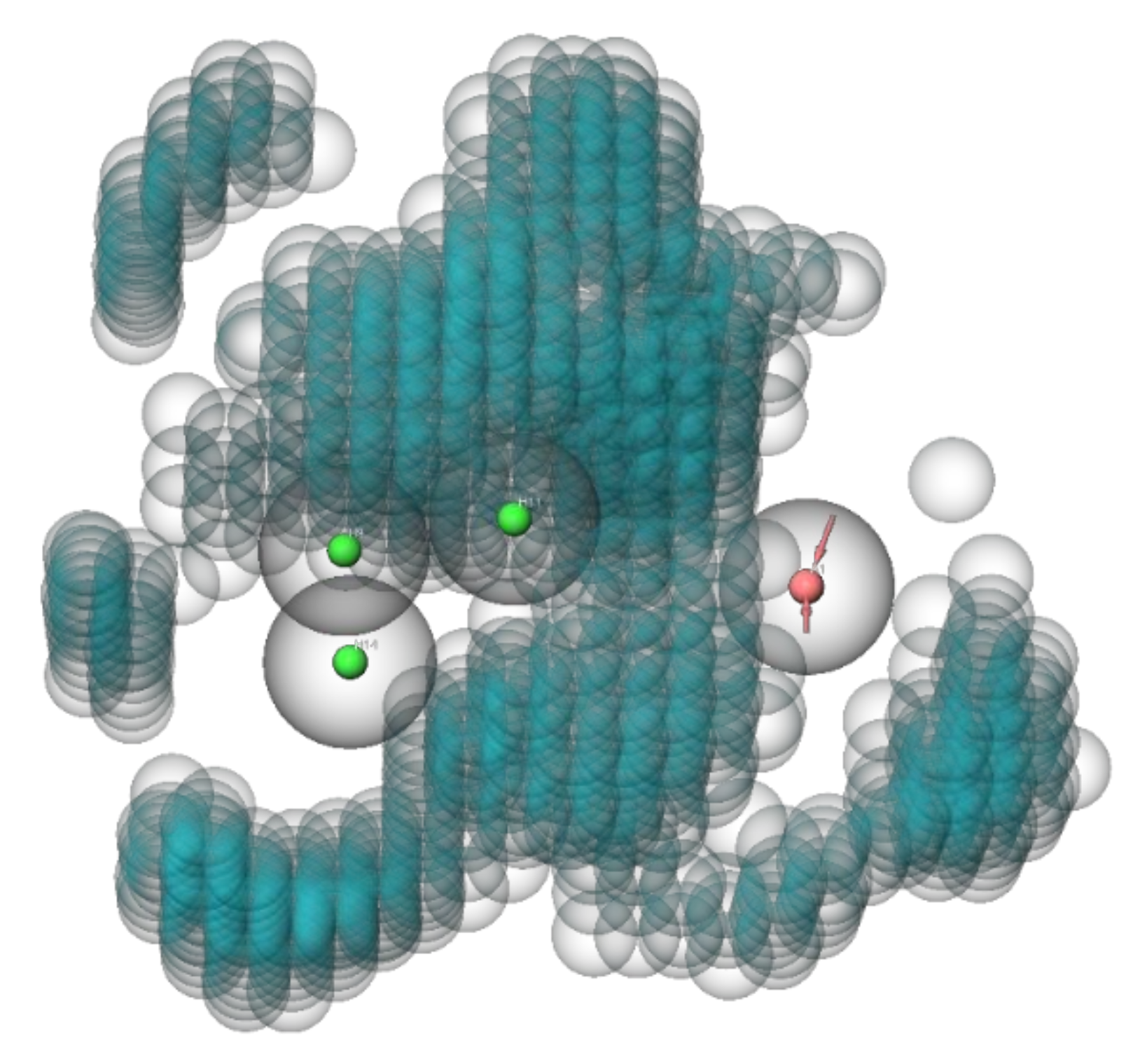
To limit the computing power for the VS with the generated QSAR model, the compounds of the ZINC
15 library were preselected by screening with an anti-preS1 activity pharmacophore model (
Figure 4). All settings for the pharmacophore hypothesis generation are described in the Material and Methods
Section 2.7. Active compounds with IC
50 < 10 µM for inhibition of [
3H]preS1-peptide binding to NTCP were used to determine features of the pharmacophore and inactive inhibitors with IC
50 > 20 µM were used to define excluding volumes. As shown in
Figure 4, the hypothesis of an anti-preS1 activity pharmacophore model revealed three hydrophobic spheres and one H-bond acceptor sphere together with clustered excluding volumes. Subsequently, ~11 million compounds of the ZINC
15 library were screened with the illustrated pharmacophore hypothesis. In addition, drug likeness filtering was applied by PHASE [
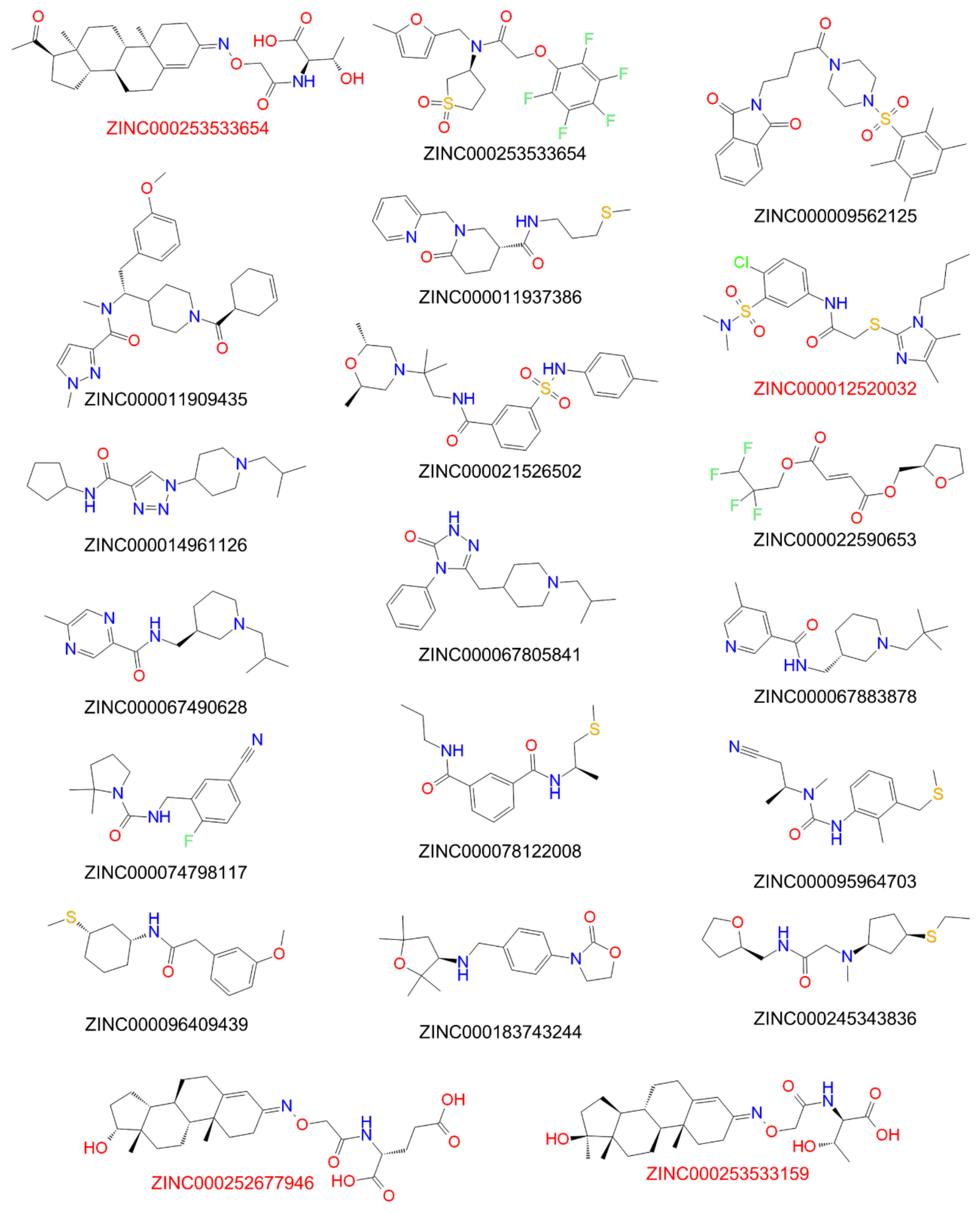
33] for this virtual screen. More than 177,000 hit compounds were identified that matched with all pharmacophore features, representing a hit rate of approximately 1.6%. These compounds then were further screened with the atom-based QSAR model resulting in a compound list with predicted anti-preS1 activities. The top 20 hits that were commercially available are listed in
Table 5 and their chemical structures are illustrated in
Figure 5. The predicted IC
50 values for [
3H]preS1-peptide binding inhibition at NTCP ranged from 7 to 16 µM (
Table 5).
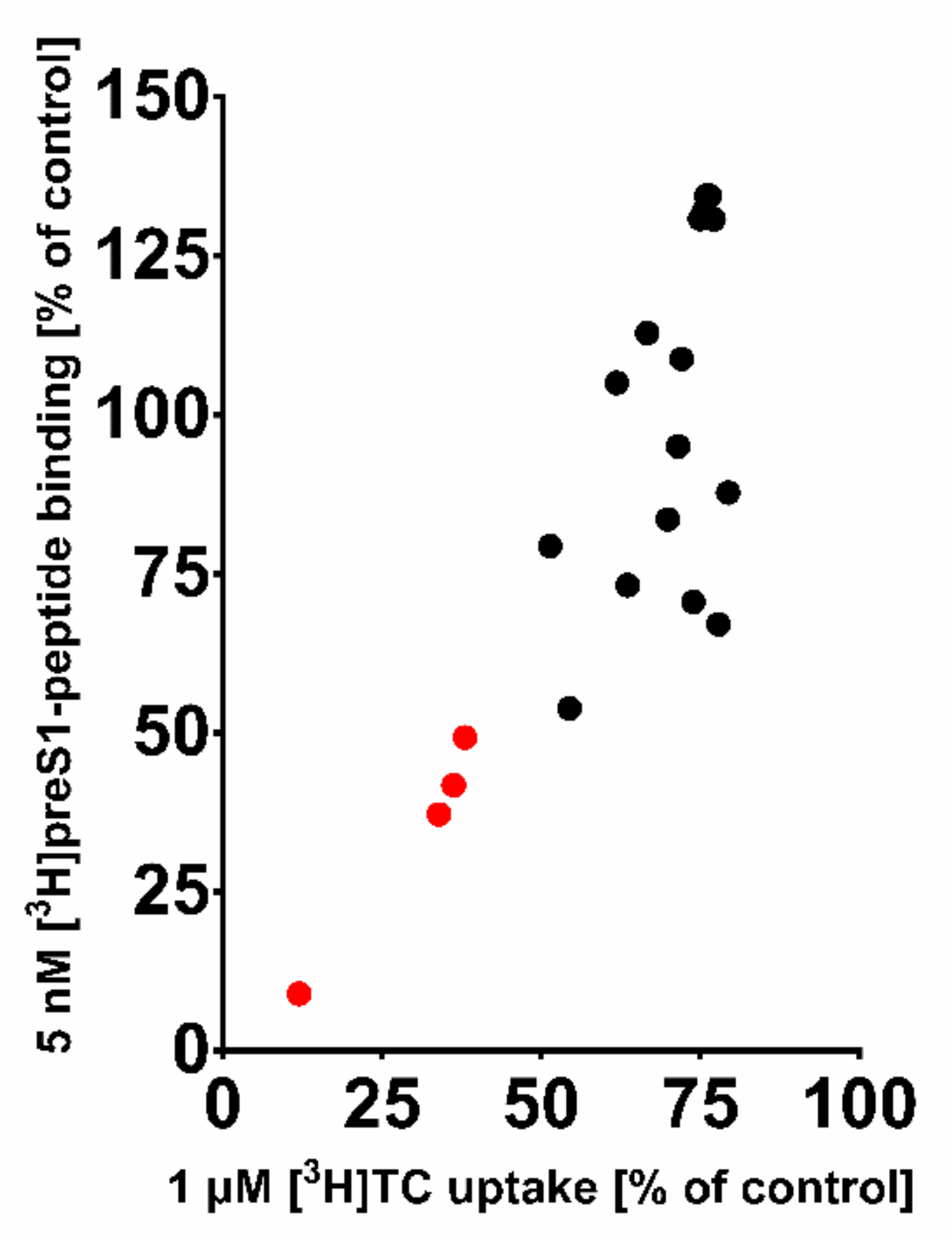
These 20 compounds were purchased and tested at 100 µM inhibitory concentrations in binding experiments with 5 nM [
3H]preS1-peptide and transport experiments with 1 µM [
3H]TC, both in NTCP-overexpressing HEK293 cells. Among this compound set only the compounds ZINC000012520032, ZINC000253533654, ZINC000253533159, and ZINC000252677946 revealed more than 50% [
3H]preS1-peptide binding inhibition (
Figure 6). Therefore, only these four compounds were further analyzed for IC
50 inhibitory concentrations (
Figure 7) and proof-of-concept HDV infection inhibition (
Figure 8). As shown in
Figure 7, all four compounds revealed concentration-dependent inhibition of [
3H]TC transport via NTCP and [
3H]preS1-peptide binding to NTCP with IC
50 ranging from 11 to 51 µM and 9 to 35 µM, respectively (
Table 6). All compounds were nearly equipotent in both inhibitory assays and, therefore, can be classified as novel non-selective NTCP inhibitors. Of note, the experimentally determined IC
50 values for [
3H]preS1-peptide binding inhibition fulfilled quite well the QSAR prediction with a deviation factor of <2.5 for all four compounds. In particular, compound ZINC000253533654 showed almost exactly the predicted activity. Summarizing that from a data set of almost 11 million chemical compounds from the ZINC
15 library, a subset of 20 compounds could be filtered out, of which four compounds indeed showed concentration-dependent inhibition of myr-preS1
2–48 lipopeptide binding. So we obtained a predictability value of approximately 20% for our two-step VS approach.
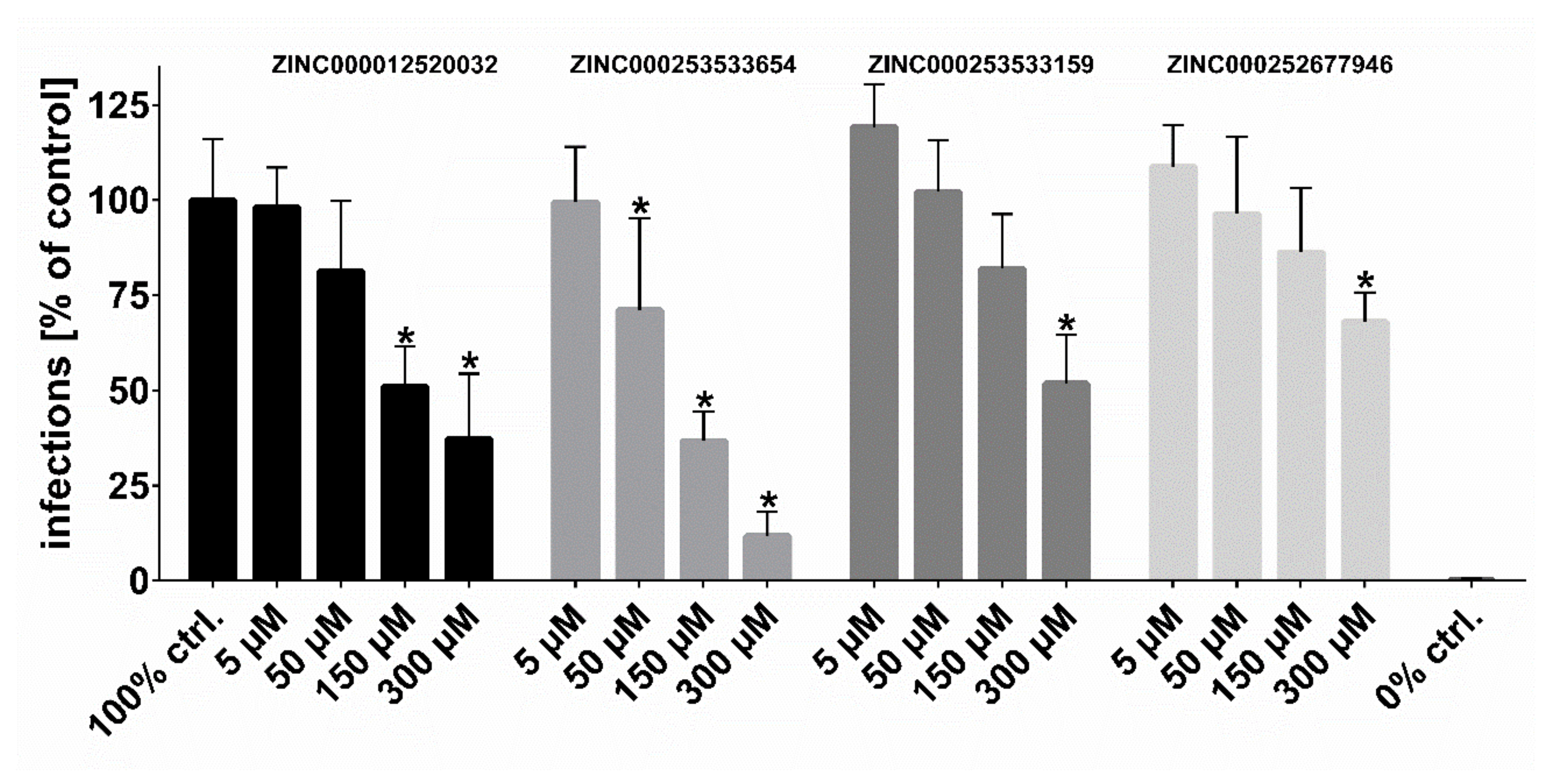
Following the workflow, the four hit compounds were experimentally validated for their inhibitory potency on in vitro HDV infection in NTCP-expressing HepG2 hepatoma cells (
Figure 8). All four compounds showed significant concentration-dependent inhibition of HDV infection with a potency rank order of ZINC000253533654 > ZINC000012520032 > ZINC000253533159 > ZINC000252677946. Cytotoxicity studies revealed no toxic effects even at highest inhibitor concentration of 300 µM over 6 h of incubation, representing the experimental conditions of the HDV infection experiments (
Figure 9).
4. Discussion
The aim of the present study was to identify novel inhibitors of NTCP with drug-like characteristics as potential therapeutics against HBV and HDV infections. HTS was not an option due to financial limitations. In addition, structure-based drug design was not possible, because no valid structural model is currently available for human NTCP. So, we decided to apply ligand-based bioinformatic methods for pharmacophore- and QSAR-guided VS of compound libraries. Our ligand-based approach is not necessarily a disadvantage, because inhibitor design solely based on crystallographic structures of the target protein can be critical due to inadequate resolution [
48] or crystallization-related artifacts of the ligand–protein complex [
49]. Furthermore, crystallographic studies may ignore discrete conformational states and anisotropic motion of the protein [
50,
51].
For ligand-based drug design, however, some aspects have to be taken into account to achieve appropriate and valid results. Data from congeneric experiments are necessary, all using the identical target, cell line, and experimental assay [
33]. Therefore, we performed inhibition studies for all compounds used for pharmacophore and QSAR modeling with the identical experimental setup. But as NTCP seems to have different substrate and inhibitor binding sites [
24], we cannot be sure that all analyzed compounds bind to the identical binding site at NTCP. This is a limitation of the approach used and a possible reason for inaccurate predictions. Also, we cannot exclude that some compounds inhibit NTCP in a competitive manner, while others may induce allosteric effects. These limitations underline the importance of the proof-of-concept in vitro HDV infection experiments that we performed with the four best-performing compounds of the present study.
The design of our inhibition and infection studies allowed us to categorize the identified inhibitors as HDV entry inhibitors acting at NTCP, as (I) their binding to NTCP was demonstrated by inhibition of [3H]TC uptake and [3H]preS1-peptide binding and (II) they were only present in the infection assay for the first 6 h of HDV exposure of the NTCP-HepG2 cells, representing the early entry phase. However, since we cannot rule out the possibility that some inhibitors might also be transported via NTCP into the HepG2 cells, additional post-entry anti-HDV effects might also be possible.
Starting from ~11 million compounds of the ZINC
15 library, we identified, in our two-step pharmacophore and QSAR VS, four out of 20 compounds that fulfilled potent [
3H]preS1-peptide binding inhibition as predicted by the QSAR model and additional proof-of-concept concentration-dependent antiviral activity in the in vitro HDV infection experiments. We thus achieved a predictability of approximately 20% for our VS system, which lays in the acceptable range of 1% to 40% for such approaches [
22]. These results indicate that the percentage of compounds that do not optimally fulfil the basic requirements for ligand-based VS is low enough in our data set to obtain a reliable prediction.
When considering the correlation of measured versus predicted activities of the test set of the generated QSAR model, a R
2 value of 0.2163 seems to be very weak. However, it is recommended not to define the accuracy of a generated QSAR model by its R
2, due to its sensitivity to the variance in the dependent variable [
52]. Furthermore, as the test set was considerably smaller than the training set, fluctuations in the R
2 value of the test set could easily occur due to fluctuations in the test set variance [
52]. The fact that the QSAR model predicted the activity of five test-set compounds out of 26 nearly exactly should be taken into consideration rather than the calculated R
2. In our VS system, we were able to reproduce this predictability of approximately 20%. In addition, hit compounds not only showed inhibitory potency to block [
3H]preS1-peptide binding to NTCP but also significantly reduced HDV infection in a concentration-dependent manner. This clearly supports the applicability of our screening system for the discovery of novel HBV/HDV inhibitors acting at NTCP. Of note, none of the hit compounds showed any cytotoxic effects on the HepG2 cell line used for infection studies, even at the highest concentrations of 300 µM. This makes these compounds attractive for further development.
Subsequent studies can be versatile. Obtained data can be used as additional input for recalculations of the generated pharmacophore and QSAR models. In addition, further hits from the top-100 list of predicted [
3H]preS1-peptide binding inhibitors can be experimentally validated and used for model optimization. Furthermore, the results of the present study can be assessed on the basis of the outcome of our previous studies with the betulin and propanolamine derivatives [
13,
14]. For both compound groups we could clearly show that only small molecular changes had significant impact on the anti-preS1 activity of the individual compounds. Furthermore, by chemical modifications we achieved a certain virus selectivity of the compounds, which is advantageous to maintaining the physiological bile acid transport function of NTCP during preS1/virus binding inhibition. As an example, the propanolamine compound A000295231 revealed a selectivity index (calculated from the mean IC
50 for transport inhibition/preS1 binding inhibition) of 65. In the case of the betulin derivatives, 3,28-di-
O-acetyl-29-hydroxybetulin revealed quite potent inhibition of the [
3H]preS1-peptide binding to NTCP, but did not inhibit the [
3H]TC transport via NTCP at all. Based on this, it would be worth generating and test sets of structural derivatives for their anti-preS1 activity. Of note, the compounds ZINC000253533654, ZINC000252677946, and ZINC000253533159 are structural homologs all based on a steroid core structure, the same as for the groups of betulin and arylmethylamino steroids. Based on this, a steroid core structure might be favorable for NTCP inhibitors.
Apart from the goal to find potent and virus-selective NTCP inhibitors for HBV/HDV entry inhibition, potent bile acid transport inhibitors acting on NTCP might also have clinical implications. As examples, patients with cholestatic liver diseases, obesity, dyslipidemia, nonalcoholic steatohepatitis, or primary biliary cholangitis could profit from hepatic bile acid uptake inhibition [
10,
53]. Based on this, the data of the present study can also be used for pharmacophore and QSAR modelling with a focus on potent bile acid inhibition irrespective of anti-preS1 activity [
24,
53].
In conclusion, the present study demonstrates, for the first time, pharmacophore and QSAR models for preS1-peptide binding inhibition at NTCP. With a two-step VS approach, novel NTCP inhibitors were identified with high prediction rate and accuracy and even demonstrated anti-HDV activity. These compounds can be used for further development of small molecular HBV/HDV entry inhibitors.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}