
Crystal Structure of a Retroviral Polyprotein: Prototype Foamy Virus Protease-Reverse Transcriptase (PR-RT)

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cloning, Expression and Purification
2.2. Selenomethionine (SeMet) Labeling
2.3. Crystallization
2.4. Data Collection and Processing
2.5. Modeling of the PFV PR Dimer Structure
2.6. Extension and Processivity Assay
2.7. Protein–DNA Cross-Linking
2.8. Accession Number(s)
3. Results
3.1. Engineering a Proteolytically Resistant Mutant of PFV PR-RT for Bacterial Expression
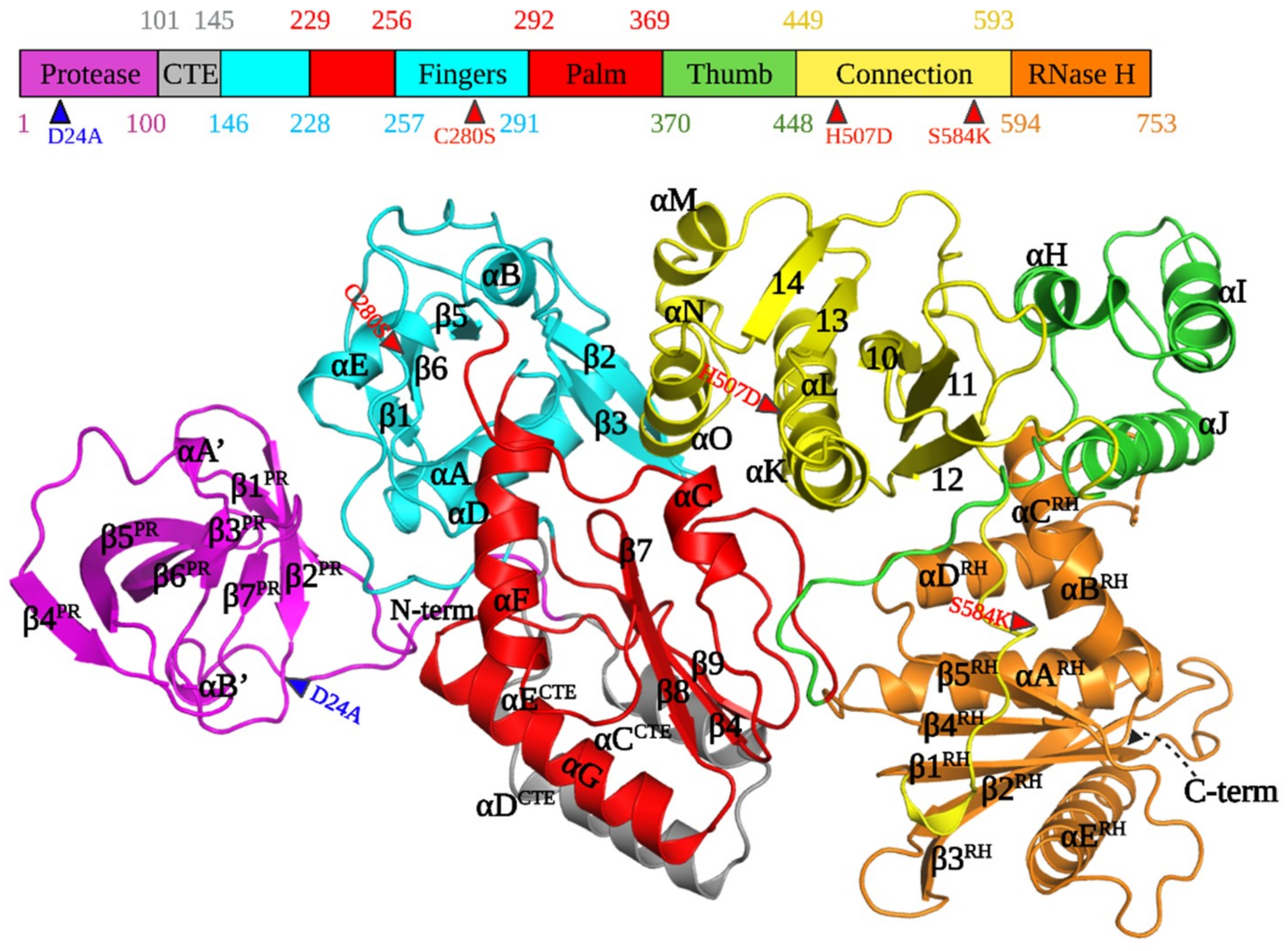
3.2. Architecture of PFV PR-RT
3.3. PR and PR-CTE
3.4. RT Domains and Subdomains
3.5. Fingers and Palm Subdomains
3.6. Thumb and Connection Subdomains
3.7. RNase H
3.8. Buried Surface Analysis of Interactions among the PR-RT Subdomains
3.9. PFV PR-RT Complexes with Nucleic Acids
4. Discussion
5. Conclusions
Importance
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hutter, S.; Mullers, E.; Stanke, N.; Reh, J.; Lindemann, D. Prototype foamy virus protease activity is essential for intraparticle reverse transcription initiation but not absolutely required for uncoating upon host cell entry. J. Virol. 2013, 87, 3163–3176. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.G.; Stenbak, C.R.; Linial, M.L. Foamy virus assembly with emphasis on pol encapsidation. Viruses 2013, 5, 886–900. [Google Scholar] [CrossRef]
- Linial, M.L. Foamy viruses are unconventional retroviruses. J. Virol. 1999, 73, 1747–1755. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.F.; Sullivan, M.D.; Linial, M.L. Evidence that the human foamy virus genome is DNA. J. Virol. 1999, 73, 1565–1572. [Google Scholar] [CrossRef] [Green Version]
- Linial, M. Why aren’t foamy viruses pathogenic? Trends Microbiol. 2000, 8, 284–289. [Google Scholar] [CrossRef]
- Baldwin, D.N.; Linial, M.L. The roles of Pol and Env in the assembly pathway of human foamy virus. J. Virol. 1998, 72, 3658–3665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, J.; Linial, M.L. Role of the foamy virus Pol cleavage site in viral replication. J. Virol. 2007, 81, 4956–4962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, G.; Yost, S.A.; Miller, M.T.; Elrod, E.J.; Grakoui, A.; Marcotrigiano, J. Structural and functional insights into alphavirus polyprotein processing and pathogenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 16534–16539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.F.; Baldwin, D.N.; Gwynn, S.R.; Yendapalli, S.; Linial, M.L. Human foamy virus replication: A pathway distinct from that of retroviruses and hepadnaviruses. Science 1996, 271, 1579–1582. [Google Scholar] [CrossRef]
- Ding, J.; Das, K.; Hsiou, Y.; Sarafianos, S.G.; Clark, A.D., Jr.; Jacobo-Molina, A.; Tantillo, C.; Hughes, S.H.; Arnold, E. Structure and functional implications of the polymerase active site region in a complex of HIV-1 RT with a double-stranded DNA template-primer and an antibody Fab fragment at 2.8 A resolution. J. Mol. Biol. 1998, 284, 1095–1111. [Google Scholar] [CrossRef] [PubMed]
- Nowak, E.; Miller, J.T.; Bona, M.K.; Studnicka, J.; Szczepanowski, R.H.; Jurkowski, J.; Le Grice, S.F.; Nowotny, M. Ty3 reverse transcriptase complexed with an RNA-DNA hybrid shows structural and functional asymmetry. Nat. Struct. Mol. Biol. 2014, 21, 389–396. [Google Scholar] [CrossRef] [Green Version]
- Tozser, J. Comparative studies on retroviral proteases: Substrate specificity. Viruses 2010, 2, 147–165. [Google Scholar] [CrossRef] [Green Version]
- Lochelt, M.; Flugel, R.M. The human foamy virus pol gene is expressed as a Pro-Pol polyprotein and not as a Gag-Pol fusion protein. J. Virol. 1996, 70, 1033–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfrepper, K.I.; Rackwitz, H.R.; Schnolzer, M.; Heid, H.; Lochelt, M.; Flugel, R.M. Molecular characterization of proteolytic processing of the Pol proteins of human foamy virus reveals novel features of the viral protease. J. Virol. 1998, 72, 7648–7652. [Google Scholar] [CrossRef] [Green Version]
- Wohrl, B.M. Structural and Functional Aspects of Foamy Virus Protease-Reverse Transcriptase. Viruses 2019, 11, 598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rethwilm, A.; Bodem, J. Evolution of foamy viruses: The most ancient of all retroviruses. Viruses 2013, 5, 2349–2374. [Google Scholar] [CrossRef]
- Katoh, I.; Ikawa, Y.; Yoshinaka, Y. Retrovirus protease characterized as a dimeric aspartic proteinase. J. Virol. 1989, 63, 2226–2232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartl, M.J.; Schweimer, K.; Reger, M.H.; Schwarzinger, S.; Bodem, J.; Rosch, P.; Wohrl, B.M. Formation of transient dimers by a retroviral protease. Biochem. J. 2010, 427, 197–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.G.; Roy, J.; Jackson, D.; Clark, P.; Boyer, P.L.; Hughes, S.H.; Linial, M.L. Foamy retrovirus integrase contains a Pol dimerization domain required for protease activation. J. Virol. 2011, 85, 1655–1661. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Smerdon, S.J.; Jager, J.; Kohlstaedt, L.A.; Rice, P.A.; Friedman, J.M.; Steitz, T.A. Structural basis of asymmetry in the human immunodeficiency virus type 1 reverse transcriptase heterodimer. Proc. Natl. Acad. Sci. USA 1994, 91, 7242–7246. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Pedersen, L.C.; Gabel, S.A.; Mueller, G.A.; Cuneo, M.J.; DeRose, E.F.; Krahn, J.M.; London, R.E. Selective unfolding of one Ribonuclease H domain of HIV reverse transcriptase is linked to homodimer formation. Nucleic Acids Res. 2014, 42, 5361–5377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Perera, L.; Mueller, G.A.; DeRose, E.F.; London, R.E. Asymmetric conformational maturation of HIV-1 reverse transcriptase. eLife 2015, 4, e06359. [Google Scholar] [CrossRef]
- Liu, H.; Naismith, J.H. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 2008, 8, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potterton, L.; Agirre, J.; Ballard, C.; Cowtan, K.; Dodson, E.; Evans, P.R.; Jenkins, H.T.; Keegan, R.; Krissinel, E.; Stevenson, K.; et al. CCP4i2: The new graphical user interface to the CCP4 program suite. Acta Cryst. D Struct. Biol. 2018, 74, 68–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Cryst. D Biol. Cryst. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Echols, N.; Headd, J.J.; Hung, L.W.; Jain, S.; Kapral, G.J.; Grosse Kunstleve, R.W.; et al. The Phenix software for automated determination of macromolecular structures. Methods 2011, 55, 94–106. [Google Scholar] [CrossRef] [Green Version]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst. D Biol. Cryst. 1997, 53, 240–255. [Google Scholar] [CrossRef]
- Boyer, P.L.; Stenbak, C.R.; Clark, P.K.; Linial, M.L.; Hughes, S.H. Characterization of the polymerase and RNase H activities of human foamy virus reverse transcriptase. J. Virol. 2004, 78, 6112–6121. [Google Scholar] [CrossRef] [Green Version]
- Rinke, C.S.; Boyer, P.L.; Sullivan, M.D.; Hughes, S.H.; Linial, M.L. Mutation of the catalytic domain of the foamy virus reverse transcriptase leads to loss of processivity and infectivity. J. Virol. 2002, 76, 7560–7570. [Google Scholar] [CrossRef] [Green Version]
- Das, K.; Martinez, S.E.; Bauman, J.D.; Arnold, E. HIV-1 reverse transcriptase complex with DNA and nevirapine reveals non-nucleoside inhibition mechanism. Nat. Struct. Mol. Biol. 2012, 19, 253–259. [Google Scholar] [CrossRef] [Green Version]
- Das, K.; Martinez, S.E.; DeStefano, J.J.; Arnold, E. Structure of HIV-1 RT/dsRNA initiation complex prior to nucleotide incorporation. Proc. Natl Acad. Sci. USA 2019, 116, 7308–7313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarafianos, S.G.; Clark, A.D., Jr.; Tuske, S.; Squire, C.J.; Das, K.; Sheng, D.; Ilankumaran, P.; Ramesha, A.R.; Kroth, H.; Sayer, J.M.; et al. Trapping HIV-1 reverse transcriptase before and after translocation on DNA. J. Biol. Chem. 2003, 278, 16280–16288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guruprasad, K.; Reddy, B.V.B.; Pandit, M.W. Correlation between stability of a protein and its dipeptide composition: A novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng. Des. Sel. 1990, 4, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Wlodawer, A.; Miller, M.; Jaskolski, M.; Sathyanarayana, B.K.; Baldwin, E.; Weber, I.T.; Selk, L.M.; Clawson, L.; Schneider, J.; Kent, S.B. Conserved folding in retroviral proteases: Crystal structure of a synthetic HIV-1 protease. Science 1989, 245, 616–621. [Google Scholar] [CrossRef]
- Sheik Amamuddy, O.; Bishop, N.T.; Tastan Bishop, O. Characterizing early drug resistance-related events using geometric ensembles from HIV protease dynamics. Sci. Rep. 2018, 8, 17938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishima, R.; Torchia, D.A.; Lynch, S.M.; Gronenborn, A.M.; Louis, J.M. Solution structure of the mature HIV-1 protease monomer: Insight into the tertiary fold and stability of a precursor. J. Biol. Chem. 2003, 278, 43311–43319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, J.; Jacobo-Molina, A.; Tantillo, C.; Lu, X.; Nanni, R.G.; Arnold, E. Buried surface analysis of HIV-1 reverse transcriptase p66/p51 heterodimer and its interaction with dsDNA template/primer. J. Mol. Recognit 1994, 7, 157–161. [Google Scholar] [CrossRef]
- Jacobo-Molina, A.; Ding, J.; Nanni, R.G.; Clark, A.D., Jr.; Lu, X.; Tantillo, C.; Williams, R.L.; Kamer, G.; Ferris, A.L.; Clark, P.; et al. Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double-stranded DNA at 3.0 A resolution shows bent DNA. Proc. Natl. Acad. Sci. USA 1993, 90, 6320–6324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohlstaedt, L.A.; Wang, J.; Friedman, J.M.; Rice, P.A.; Steitz, T.A. Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science 1992, 256, 1783–1790. [Google Scholar] [CrossRef] [Green Version]
- Champoux, J.J.; Schultz, S.J. Ribonuclease H: Properties, substrate specificity and roles in retroviral reverse transcription. FEBS J. 2009, 276, 1506–1516. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Sawyer, N.; Regan, L. Protein-protein interactions: General trends in the relationship between binding affinity and interfacial buried surface area. Protein Sci. 2013, 22, 510–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chothia, C. The nature of the accessible and buried surfaces in proteins. J. Mol. Biol. 1976, 105, 1–12. [Google Scholar] [CrossRef]
- Krissinel, E. Stock-based detection of protein oligomeric states in jsPISA. Nucleic Acids Res. 2015, 43, W314–W319. [Google Scholar] [CrossRef] [Green Version]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Huang, H.; Harrison, S.C.; Verdine, G.L. Trapping of a catalytic HIV reverse transcriptase*template:primer complex through a disulfide bond. Chem. Biol. 2000, 7, 355–364. [Google Scholar] [CrossRef] [Green Version]
- Harris, K.S.; Reddigari, S.R.; Nicklin, M.J.; Hammerle, T.; Wimmer, E. Purification and characterization of poliovirus polypeptide 3CD, a proteinase and a precursor for RNA polymerase. J. Virol. 1992, 66, 7481–7489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yost, S.A.; Marcotrigiano, J. Viral precursor polyproteins: Keys of regulation from replication to maturation. Curr. Opin. Virol. 2013, 3, 137–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Selenomethionine (SeMet) PFV PR-RT (D24A+CSH) Mutant) | PFV PR-RT WT (D24A) | |
|---|---|---|
| PDB ID | 7KSE | 7KSF |
| Wavelength (Å) | 0.98 | 0.98 |
| Resolution range (Å) | 50.0–3.0 (3.18–3.0) | 38.03–2.9 (3.004–2.9) |
| Space group | C2 | C2 |
| Unit cell (Å/deg) | 240.38, 53.35, 74.92/90, 100, 90 | 228.21, 52.54, 75.43/90, 90.09, 90 |
| Unique reflections | 19100 (2990) | 19443 (1752) |
| Multiplicity | 30.4 (29.7) | 5.1 (5.1) |
| Completeness (%) | 99.7 (98.0) | 96.1 (88.4) |
| Mean I/sigma(I) | 10.6 (2.7) | 7.1 (1.0) |
| Wilson B-factor (Å2) | 70.3 | 92.1 |
| Rmerge | 0.331 (1.973) | 0.128 (1.532) |
| Rmeas | 0.337 (2.007) | 0.157 (1.888) |
| Rpim | 0.061 (0.367) | 0.064 (0.812) |
| CC1/2 | 0.996 (0.818) | 0.993 (0.334) |
| Rwork | 0.277 (0.382) | 0.224 (0.370) |
| Rfree | 0.312 (0.376) | 0.258 (0.381) |
| Number of non-hydrogen atoms | 5911 | 5915 |
| macromolecules | 5898 | 5905 |
| ligands | 1 | 1 |
| solvent | 12 | 9 |
| Protein residues | 743 | 743 |
| RMS bonds (Å) | 0.007 | 0.004 |
| RMS angles (deg) | 1.40 | 0.71 |
| Ramachandran favored (%) | 89.40 | 94.43 |
| Ramachandran allowed (%) | 10.05 | 5.30 |
| Ramachandran outliers (%) | 0.54 | 0.27 |
| Rotamer outliers (%) | 0.46 | 0.77 |
| Clash score | 23.68 | 11.65 |
| Average B-factor (Å2) | 85.66 | 103.78 |
| macromolecules | 85.71 | 103.78 |
| ligands | 110.53 | 123.65 |
| solvent | 62.54 | 103.14 |
| PR | PR-CTE | Fingers | Palm | Thumb | Connection | RNase H | |
|---|---|---|---|---|---|---|---|
| PR | - | 338 (−4.8) | 632 (−7.5) | - | - | - | - |
| PR-CTE | 338 (−4.8) | - | 257 (1.0) | 1009 (−17.4) | - | - | - |
| Fingers | 632 (−7.5) | 257 (1.0) | - | 785 (−7.6) | - | 212 (−1.7) | - |
| Palm | - | 1009 (−17.4) | 785 (−7.6) | - | 359 (0.0) | 547 (−7.2) | |
| Thumb | - | - | - | 359 (0.0) | - | 1216 (−17.9) | 164 (−2.0) |
| Connection | - | - | 212 (−1.7) | 547 (−7.2) | 1216 (−17.9) | - | 425 (−3.3) |
| RNase H | - | - | - | - | 164 (−2.0) | 425 (−3.3) | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harrison, J.J.E.K.; Tuske, S.; Das, K.; Ruiz, F.X.; Bauman, J.D.; Boyer, P.L.; DeStefano, J.J.; Hughes, S.H.; Arnold, E. Crystal Structure of a Retroviral Polyprotein: Prototype Foamy Virus Protease-Reverse Transcriptase (PR-RT). Viruses 2021, 13, 1495. https://doi.org/10.3390/v13081495
Harrison JJEK, Tuske S, Das K, Ruiz FX, Bauman JD, Boyer PL, DeStefano JJ, Hughes SH, Arnold E. Crystal Structure of a Retroviral Polyprotein: Prototype Foamy Virus Protease-Reverse Transcriptase (PR-RT). Viruses. 2021; 13(8):1495. https://doi.org/10.3390/v13081495
Chicago/Turabian StyleHarrison, Jerry Joe E. K., Steve Tuske, Kalyan Das, Francesc X. Ruiz, Joseph D. Bauman, Paul L. Boyer, Jeffrey J. DeStefano, Stephen H. Hughes, and Eddy Arnold. 2021. "Crystal Structure of a Retroviral Polyprotein: Prototype Foamy Virus Protease-Reverse Transcriptase (PR-RT)" Viruses 13, no. 8: 1495. https://doi.org/10.3390/v13081495