1. Introduction
Kaposi Sarcoma Herpesvirus (KSHV) is an oncogenic herpesvirus and one of only two human herpesviruses known to infect B lymphocytes. KSHV lymphotropism results in two lymphoproliferative disorders, PEL and MCD [
1,
2]. Despite decades of research related to KSHV pathogenesis, essential questions remain unanswered related to how KSHV invades a new human host and particularly how the virus invades the human immune system. Studies addressing these gaps are particularly critical to inform the rational development of prophylactic vaccine strategies to limit KSHV transmission.
The highly conserved glycoproteins gB, gH and gL form the canonical core entry machinery for all herpesviruses, and various additional glycoproteins, which vary between herpesviruses, work together with these core proteins to mediate the broad cellular tropism characteristic of human herpesviruses [
3]. There is a considerable body of literature characterizing the cellular receptors and KSHV glycoproteins involved in viral entry in vitro. However, no study to date has succeeded in completely abrogating KSHV infection via inhibition of a specific cellular receptor, suggesting that KSHV universally employs redundant entry mechanisms involving multiple cellular receptors and glycoproteins [
4].
In epithelial, endothelial and fibroblast cell types, KSHV infection is facilitated by attachment of gB, gH/gL and/or K8.1 to heparin sulfate proteoglycans, however our recent work showed that HSPG play no role in infection of primary tonsil-derived B lymphocytes [
5]. Similarly, a previous study from our group recently showed that KSHV-gH is essential for entry into all adherent cell types tested, but is not required for entry into the MC116 lymphoma cell line [
6]. Thus, there is accumulating evidence that KSHV uses different mechanisms for entry into B cells compared to adherent cells.
DC-SIGN, also known as, Dendritic Cell-Specific ICAM-3-Grabbing Non-integrin, CD209, is a type II C-type (calcium-dependent) lectin receptor [
7] and is highly expressed in cells of the immune system such as dendritic cells (DCs), in dermal and mucosal tissues, monocytes, macrophages, B lymphocytes from peripheral blood, and tonsillar B lymphocytes [
8,
9]. DC-SIGN plays a critical role in the innate and adaptive immune response by facilitating immune cell signaling, adhesion and migration [
10]. DC-SIGN is utilized as an entry receptor by many viruses such as Ebola virus, HIV-1, Hepatitis C, KSHV, SARS coronavirus, and Dengue virus [
11]. Studies have demonstrated that KSHV uses DC-SIGN as an entry receptor in dendritic cells and macrophages (Rappocciolo, 2006 #1195), and neutralization of DC-SIGN was previously shown to inhibit KSHV infection of activated blood-derived and resting tonsil-derived B lymphocytes [
8]. KSHV-gB binds to DC-SIGN in a dose dependent manner, but this interaction has yet to be deemed essential for KSHV entry in any cell type [
12].
In this study, our primary objectives are to evaluate the role of KSHV-gH in entry into primary B lymphocytes, and determine whether DC-SIGN is the primary receptor for KSHV in B lymphocytes as has been previously reported [
8]. Finally, we seek to examine whether either factor (DC-SIGN as a receptor and KSHV-gH as a glycoprotein) dictates the susceptibility of any specific B cell subsets to KSHV infection. Our results demonstrate that both DC-SIGN and gH are dispensable for infection of tonsil-derived B lymphocytes and suppression of these factors does not substantially alter KSHV tropism in B lymphocytes. Interestingly, by working with these two factors in combination, we are able to establish that KSHV has two mechanisms for entry into the centrocyte subset: one dependent on gH and the other dependent on DC-SIGN. Taken together, our results reveal that mechanisms of KSHV entry into B lymphocytes are complex, multi-factoral and subset-specific.
2. Materials and Methods
2.1. Preparation of Cell-Free Recombinant KSHV Virions
iSLK cell lines harboring BAC16-KSHV-WT-eGFP [
13] and BAC16-KSHV-∆gH-eGFP [
6] were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% Cosmic Calf Serum (CCS), PSG, puromycin (1 µM), G418 (250 µg/mL), and hygromycin (1.2 mg/mL) at 37 °C in 5% CO
2. For virus preparations, 12 × T185 flasks at 80–90% confluence were stimulated for 72 h with 3 mM sodium Butyrate and 2 µM doxycycline hyclate. At 3 days post induction, supernatants were clarified by centrifugation at 1700 rpm for 12 min at 4 °C and filtered with a 0.45 µm vacuum filter. Virions were pelleted out of clarified supernatant over 25% sucrose in TNE (50 mM Tris [pH 7.4], 100 mM NaCl, 0.1 mM EDTA, pH 7.4) by centrifugation at 22,000 rpm for 2 h. Virus pellets were resuspended in 2 mL TNE and stored at −80 °C. Infectious titer doses were determined for iSLK-BAC16-KSHV-WT-eGFP by serial dilution infection on human fibroblasts and quantified at 3 days post infection via flow cytometry. Infectious titer doses were determined for iSLK-BAC16-KSHV-∆gH-eGFP by calculating equal genome copy number to that of iSLK-BAC16-KSHV-WT-eGFP via quantitative PCR (qPCR).
2.2. Isolation of Primary Lymphocytes from Human Tonsil
De-identified human tonsil specimens were obtained from the National Disease Research Interchange (NDRI) following routine tonsillectomies. Less than 24 h post-surgery, tonsil specimens were shipped and delivered in DMEM+PSG to the laboratory. Primary lymphocytes were extracted via dissection and maceration of tonsil tissues in RPMI media. Lymphocyte-containing media were passed through a 40 µm filter, and pelleted at 1500 rpm for 5 min. Red blood cell lysing solution (0.15 M ammonium chloride, 10 mM potassium bicarbonate, 0.1 M EDTA) was utilized to lyse red blood cells present in the lymphocyte preparation. Following 3 min of RBC lysis, lymphocytes were diluted in 50 mL of PBS, manually counted and pelleted at 1500 rpm for 5 min. Aliquots of 1 × 108 cells were resuspended in 1 mL of freezing media (90% FBS, 10% DMSO) and cryopreserved.
2.3. Isolation and KSHV Infection of Total B lymphocytes
Tonsil primary lymphocyte suspensions were thawed at 37 °C, slowly diluted to 5 mL with RPMI, and pelleted at 1500 rpm for 5 min. Pellets were resuspended in 1 mL RPMI with 20% FBS, 100 µg/mL DNase I, and 100 µg/mL Primocin. Cells were maintained in a low-binding 24 well plate at 37 °C and 5% CO2 incubator for two hours. After recovery, total lymphocytes were counted and total B cells were isolated using Mojosort™ Human Pan B cell isolation kit (Biolegend 480082) according to manufacturer’s instructions. Bound cells that were non-B cells were retained and maintained in 1 mL RPMI, 20% FBS at 37 °C and 5% CO2 incubator.
Recombinant KSHV virion preparations were titrated on human fibroblasts and an ID20 was calculated by linear regression. The equivalent ID20 dose from fibroblasts (in µL/cell) was diluted in serum free RPMI media and used to infect 1 million B cells. For experiments including KSHV-∆gH, the WT virus was used at ID20 doses as described above, and KSHV-∆gH was used at an equivalent genome dose based on the WT stock. In all experiments, Mock-infected cultures were included as an internal reference for the GFP positive signal and to allow analysis of culture-specific effects. B cells in infection media were spinoculated at 1000 rpm for 30 min at 4 °C in 12 × 75 mm round bottom tubes. After spinoculation, tubes were incubated at 37 °C for an additional 30 min. After incubation, infected cultures were transferred to X-ray irradiated CDW32 L cells in a 48 well plate and reconstituted with 20% fetal bovine serum, 100 µg/mL of Primocin and 1 million cells from the bound fraction of the B cell isolation for a final concentration of 4 million cells/mL. Reconstituted lymphocyte cultures were incubated at 37 °C, 5% CO2 for the duration of the experiment.
2.4. DC-SIGN Neutralization
Total B lymphocytes were isolated as in 2.3 and were incubated with Human DC-SIGN/CD209 Antibody [anti-DC-SIGN mAb] (R&D systems MAB161-100) at varying concentrations (0 µg/mL, 2.5 µg/mL, 5 µg/mL) for 30 min on ice prior to infection as described above.
2.5. DC-SIGN Depletion
Total B lymphocytes were isolated as described above, and were further separated into DC-SIGN+ (bound) and DC-SIGN- (unbound) fractions using CD209 (DC-SIGN) MicroBeads (Miltenyi 130-092-868) according to manufacturer’s instructions. Following separation, half of the DC-SIGN- B cells were reconstituted with DC-SIGN+ B cell fraction and the remaining cultures remained depleted. These depleted and reconstituted samples were Mock-infected or infected with KSHV-WT and KSHV-∆gH and cultured as described above.
2.6. Flow Cytometry Analysis
At day 0 (baseline) or at 3 days post-infection (3 dpi), 5 × 105 lymphocytes were aliquoted into a 96-well round bottom plate and pelleted at 1500 rpm for 5 min. Resuspension of the pellet was performed with 100 µL PBS containing zombie violet fixable viability stain (Biolegend Cat# 423113) and incubated on ice for 15 min. After incubation, 100 µL PBS, containing 2% FBS and 0.5% BSA and 0.1% sodium azide (FACS Block) was added to the wells. Cells were pelleted at 1500 rpm for 5 min and resuspended in 200 µL FACS Block followed by a 10 min incubation on ice. Cells were pelleted at 1500 rpm for 5 min and resuspended in 50 µL of PBS containing 0.5% BSA and 0.1% sodium azide (FACS Wash), 10 µL BD Brilliant Stain Buffer Plus (BD 566385) and antibodies as follows: IgD-BUV395 (2.5 µL/test BD 563823), CD77-BV510 (2.0 µL/test BD 563630), CD138-BV650 (2 µL/test BD 555462), CD27-BV750 (2 µL/test BD 563328), CD19-PerCPCy5.5 (2.0 µL/test BD 561295), CD38-APC (10 µL/test BD 560158), CD20-APCH7 (2 µL/test BL 302313), and DC-SIGN- PE-Cy7 (2 µL/test BD 330114) and incubated on ice for 15 min. After incubation, 150 µL FACS Wash was added. Cells were pelleted at 1500 rpm for 5 min followed by two washes with FACS Wash. Cells were collected in 200 µL FACS Wash for flow cytometry analysis. Sample data and appropriate compensation controls were acquired on a BD Fortessa X20 flow cytometer and analyzed using FlowJo Software.
2.7. RT-PCR Analysis for Viral Transcripts
At 3 days post infection, 2 × 10
6 lymphocytes were harvested into Trizol and an equal volume of DNA/RNA shield (Zymo Research R110-250, Irvine, CA, USA) was added. RNA extraction was performed using Zymo Directzol Microprep (Zymo Research R2060) according to manufacturer instructions. RNA was eluted in 10 µL H
2O containing 2U RNase inhibitors and a second DNase step was performed for 30 min using the Turbo DNA-Free kit (Invitrogen AM1907M, Waltham, MA, USA) according to manufacturer instructions. Nested RT-PCR amplicon for GAPDH was designed based on NCBI gene id 2597 and LANA and K8.1 assays were designed based on the BAC16 reference sequence (Genbank MK733609.1) One-step RT-PCR cDNA synthesis and preamplification of GAPDH, LANA and K8.1 transcripts was performed on 15 ng of total RNA using the Superscript III One-step RT-PCR kit (ThermoFisher 12574026, Waltham, MA, USA) and 2 µM outer primers for each target gene (
Table 1). Duplicate no RT (NRT) control reactions were assembled for each sample containing only Platinum Taq DNA polymerase (Thermofisher 15966005) instead of the Superscript III RT/Taq DNA polymerase mix. After cDNA synthesis at 50 °C for 15 min 20 cycles of target pre-amplification was performed with an annealing temperature of 60 °C and 30second extension at 68 °C. Then, 2 µL of pre-amplified cDNA or NRT control reaction was used as template for multiplexed real-time PCR reactions using TaqProbe 5× qPCR MasterMix -Multiplex (ABM MasterMix-5PM), 5% DMSO, primers at 900 nM and probes at 250 nM against target genes (
Table 1) and analyzed using a standard 40 cycle program on a Biorad real time thermocycler. Data is represented as quantitation cycle (Cq) and assays in which there was no detectable Cq value were set numerically as Cq = 41 for analysis and data visualization.
2.8. Statistical Analysis
Data plots and statistical analysis were performed in Rstudio software (version 7.0) using reshape2 [
14], and tidyverse [
15] packages. Statistical analysis was performed using R package: rstatix [
16]. Specific statistical tests and the resulting values are described in detail in the corresponding figure legends.
4. Discussion
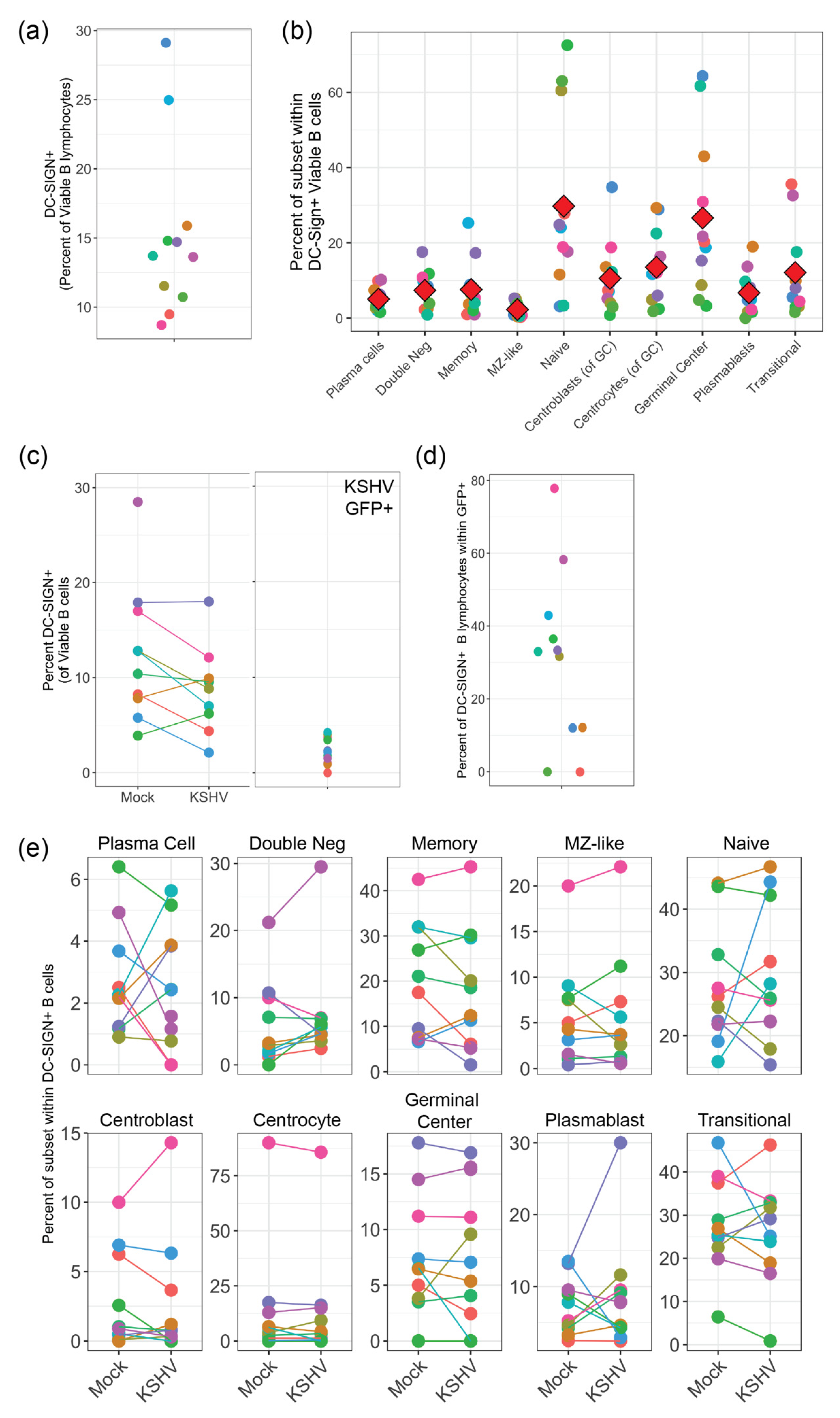
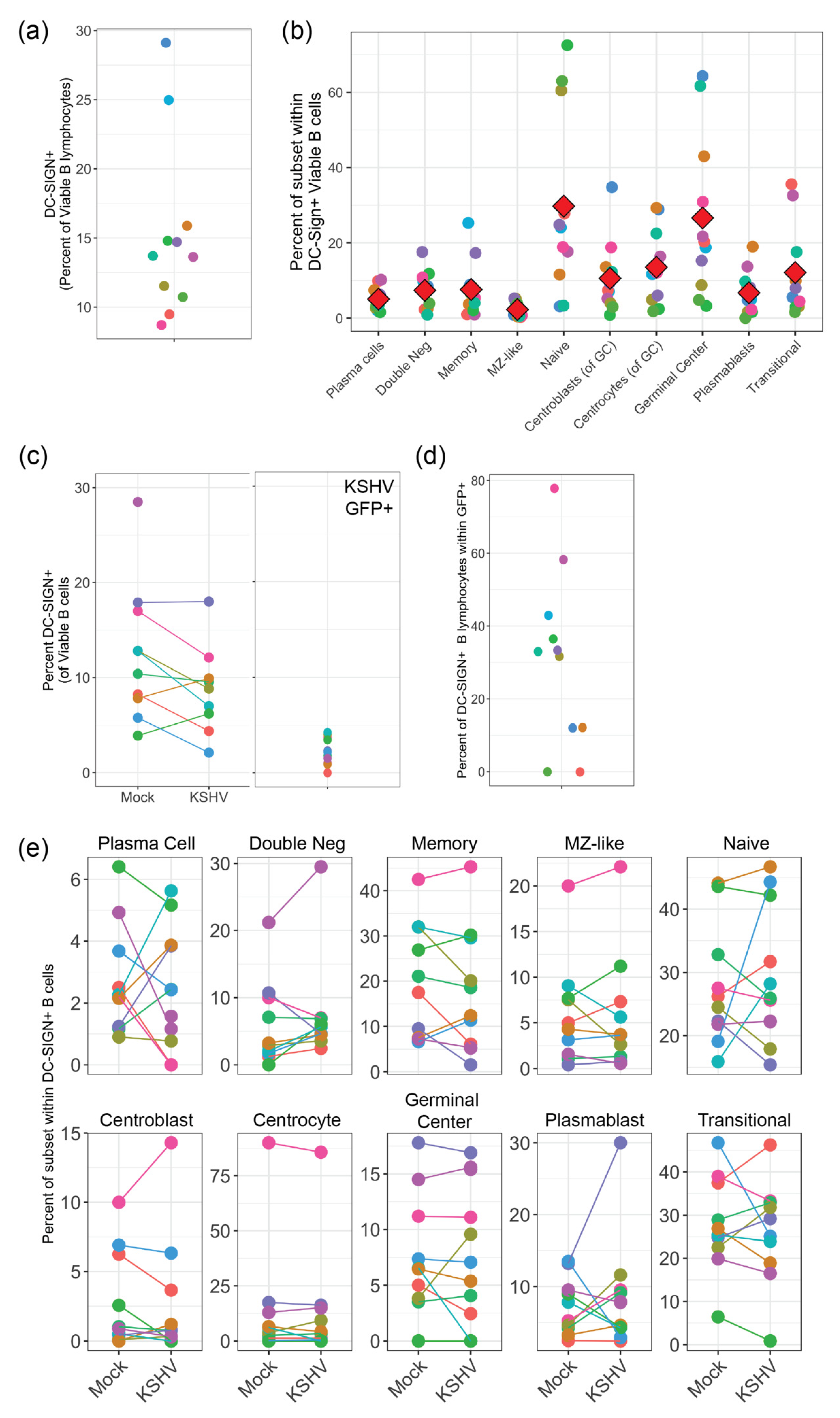
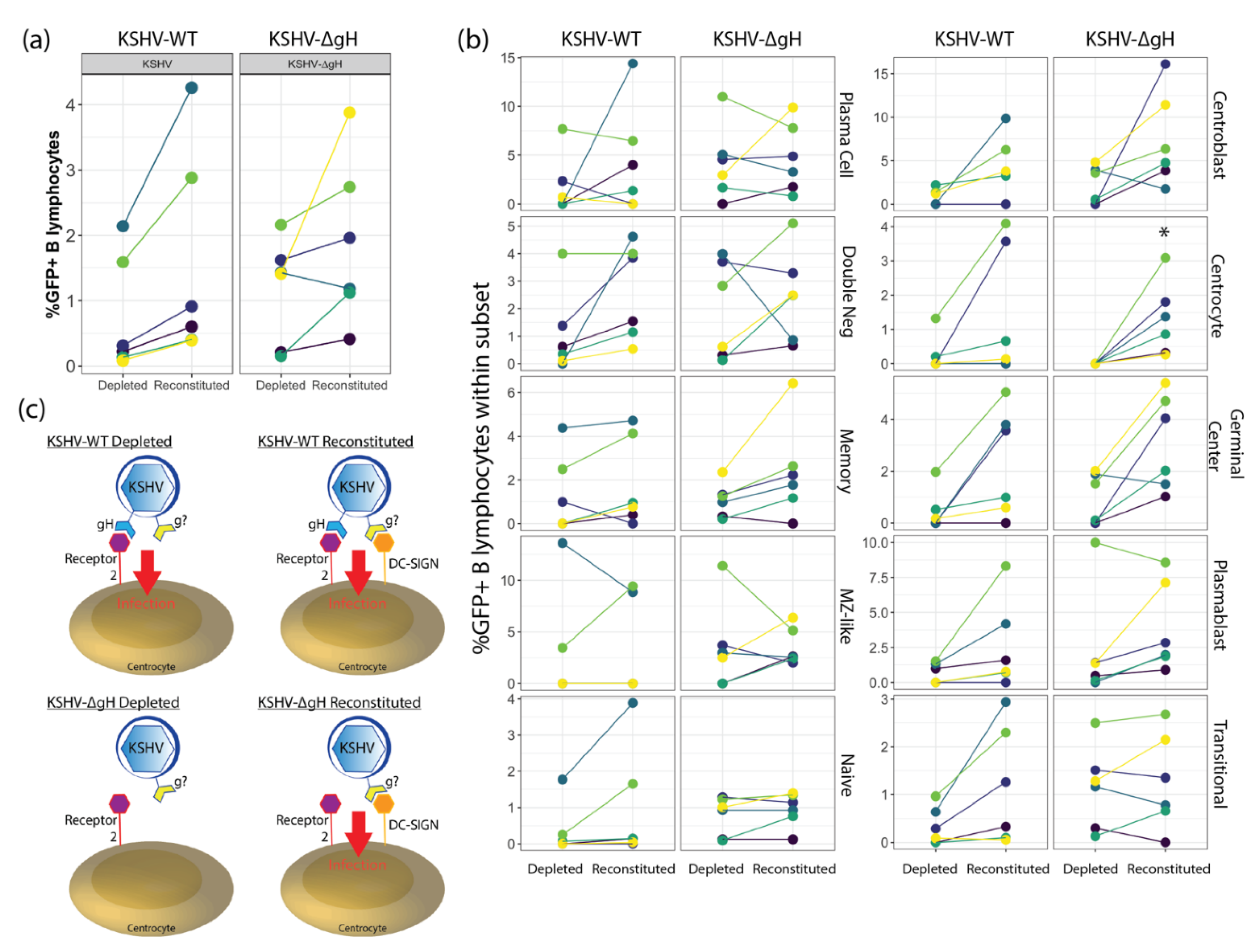
Our results presented in this study indicate that KSHV can use DC-SIGN as an attachment/entry factor, but DC-SIGN is not required for entry into any B cell subsets. In particular, depletion of DC-SIGN-expressing B cells had a more profound effect than antibody-mediated neutralization of DC-SIGN. Indeed, KSHV infection was overall increased in our neutralization experiments. These results contrast significantly with the only previous study to perform an in-depth analysis on the role of DC-SIGN as an entry receptor for KSHV, which was performed by Rappocciolo et al. in 2008 [
8]. This particular study utilized both unstimulated tonsil-derived B cells and peripheral blood B-cells that were activated with CD40 ligand (CD40L) and interleukin 4 (IL-4) as target cells and wild-type KSHV virions derived from BCBL-1 PEL cells as virus inoculum. In our results, we show a lower percentage of DC-SIGN+ B cells compared to this previous study possibly indicating that our tonsil B cells are less activated. One reason for this discrepancy may be methodological. We avoid using Ficoll-Hypaque and similar agents for lymphocyte purification and we utilize negative selection strategies for B cell isolation in order to avoid aberrant activation of cells in our system. Importantly, we utilized multi-color flow cytometry for our analysis method which allows us to see KSHV infection using the constitutively expressed GFP reporter present in the BAC16 genome (which we have previously shown is a reliable marker for KSHV tropism [
5]) alongside surface antigen markers for B cell subsets. These techniques were distinct from the previous study which analyzed B cells as a single population and utilized virus detection methods that were biased towards lytic replication as opposed to quantitating infected cell numbers.
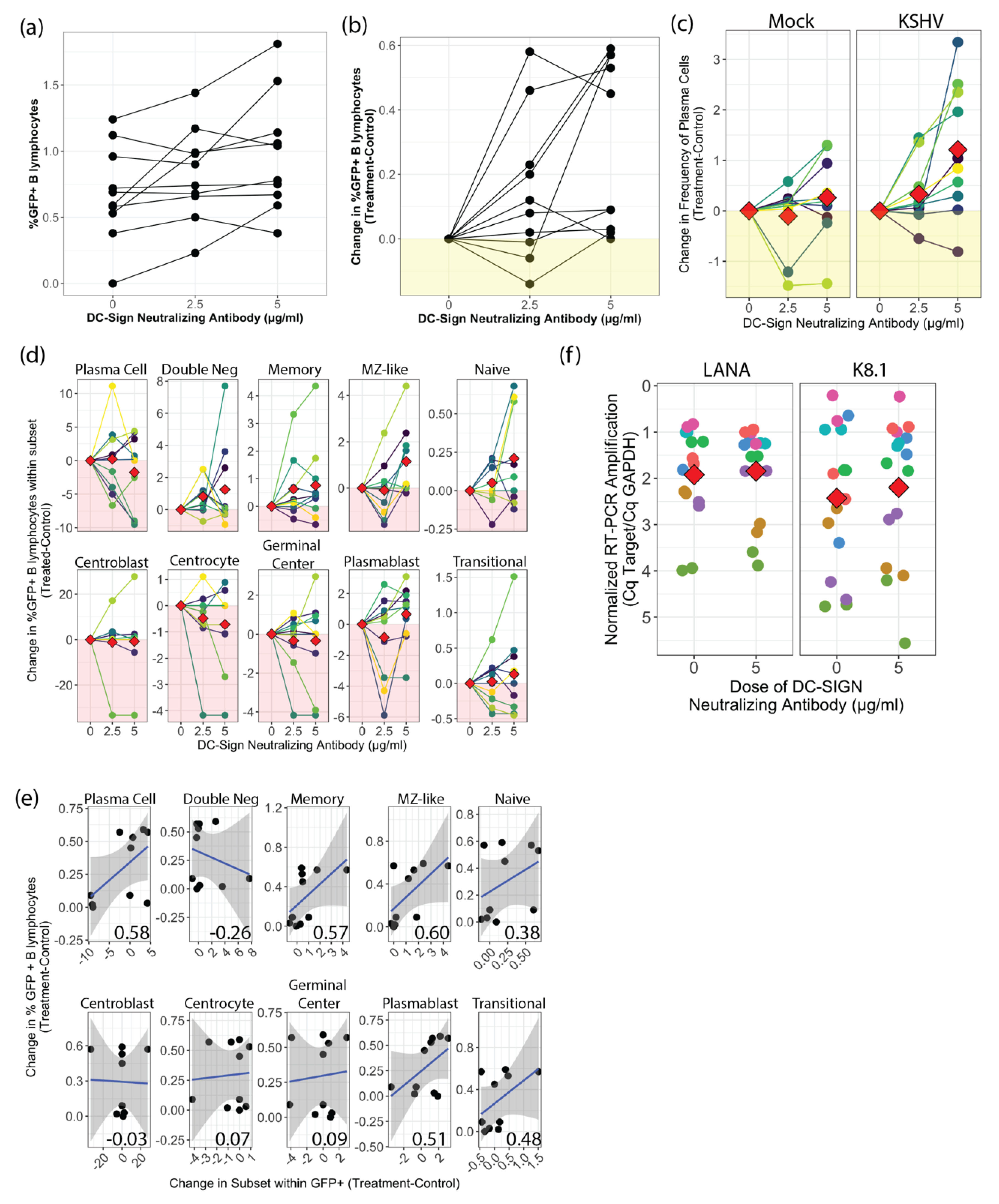
When we employed a neutralizing antibody to disrupt any interaction between DC-SIGN and KSHV glycoproteins, we observed significantly increased KSHV infection at the highest dose (5 µg/mL), which was also the optimal dose for inhibition of DC-SIGN - ICAM3 interaction during characterization of the neutralizing antibody by the manufacturer. These results differ from those reported in Rappocciolo et al., where use of the same anti-DC-SIGN neutralizing antibody clone but at much higher concentrations (20 µg/mL) effectively blocked KSHV infection of activated peripheral B-cells [
8]. Aside from the differing mAb concentrations, our culture model recapitulates the total tonsil lymphocyte environment after infection. Thus, we might hypothesize this mAb enhancement effect may be mediated by activation of DC-SIGN signaling altering other attachment factors or via altered interactions involving non-B cells in the culture model. Our analysis of the distribution of KSHV infection within B-cell subsets did not reveal any change in KSHV targeting of B-cells with DC-SIGN neutralization, and RT-PCR results show no increase in lytic replication in culture where the neutralizing antibody was used. Thus, our data provide no evidence that the increase in overall infection with DC-SIGN neutralization is a result of altered KSHV targeting or spread. We did observe an increase in total plasma cell numbers in DC-SIGN neutralized, KSHV-infected lymphocyte cultures. Our previous work has shown that our lymphocyte culture system does not favor the survival of plasma cells, but KSHV infection increases overall plasma cell numbers at 3 dpi [
5]. Recently, activation of DC-SIGN signaling via antibody binding was shown to promote survival of B lymphoma cells [
18]. Thus, the combination of DC-SIGN neutralization and KSHV infection may synergistically promote the survival of plasma cells in our culture system, and we speculate that the presence of plasma cells promotes KSHV infection via unknown mechanisms that are possibly related to the cytokine milieu. Overall, our results collectively show that DC-SIGN is not required for KSHV entry into any B-cell subset.
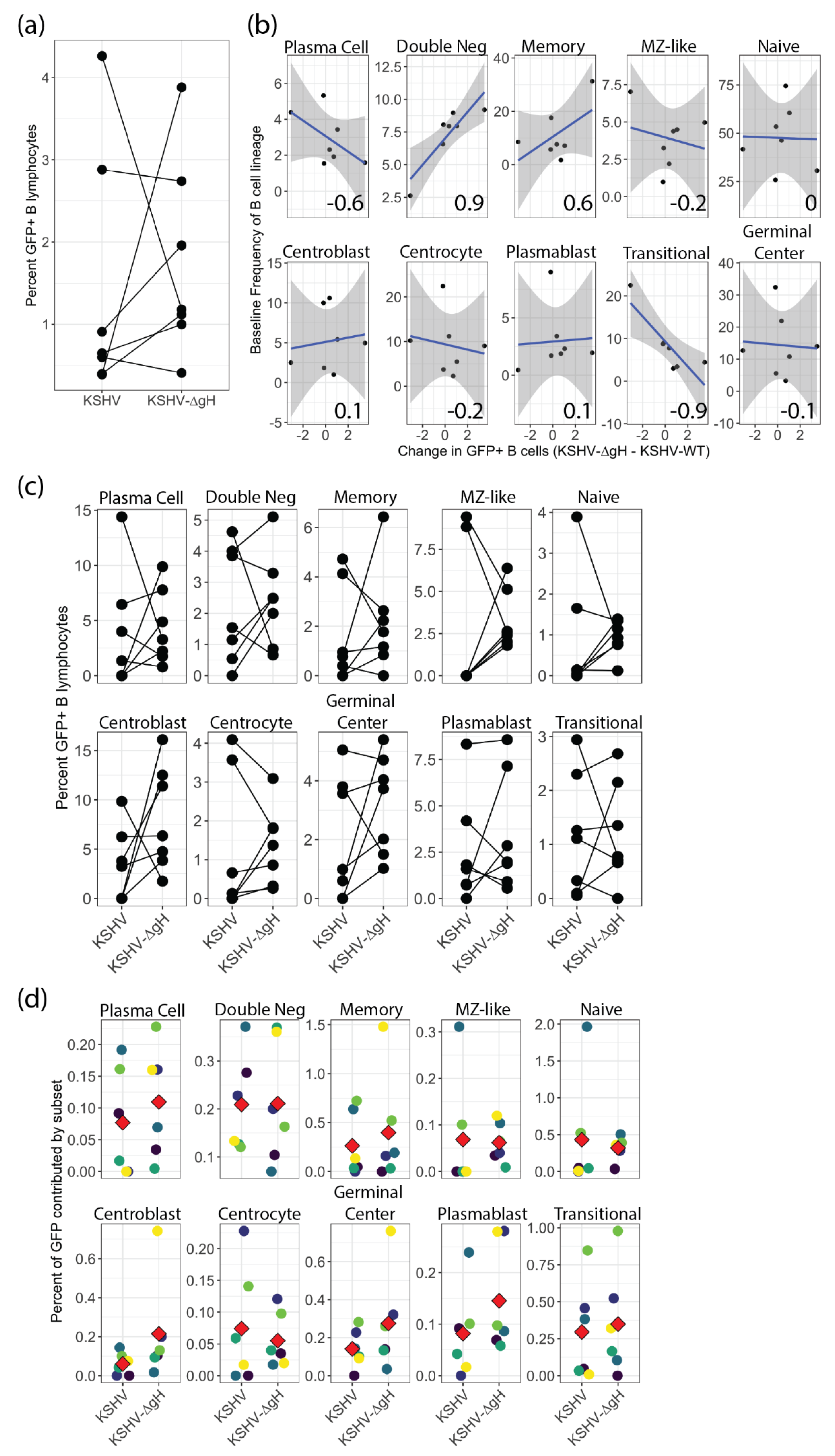
To date, it is unknown which viral glycoproteins are essential for KSHV entry in tonsil derived B lymphocytes, and this gap in our understanding limits the rational design of vaccines designed to limit the ability of KSHV to invade the immune system. We recently showed that gH is essential for KSHV entry into epithelial cells, endothelial cells and fibroblasts but is not required for entry into the MC116 B-cell lymphoma cell line [
6]. Our current study extends these findings and shows that gH is also not required for entry into primary tonsil lymphocytes. Interestingly, our results revealed tonsil donor-specific differences in the infectivity of KSHV-∆gH compared to KSHV-WT, and that these differences are correlated to the frequencies of transitional and double negative B cells in the original tonsil sample. These results provide a critical insight indicating that vaccination strategies concentrated on KSHV-gH may leave the immune system vulnerable to infection. However, our DC-SIGN depletion experiments show that there is a gH-dependent mechanism for KSHV entry into centrocytes. Thus, although gH is not strictly required for entry into any B cell type, gH-dependent entry mechanisms do exist for tonsil-derived B cells. Taken together, our results indicate that KSHV entry in B lymphocytes is complex, multi-factoral and that subset-dependent entry mechanisms exist but, in all cases, KSHV has multiple entry points to exploit on any given B cell subset.
Our study does have some limitations that require further research. In particular, for the neutralization experiment we chose to utilize a neutralizing antibody that may not be optimal for the inhibition of DC-SIGN and KSHV glycoprotein interactions. The anti-DC-SIGN antibody we utilized is optimized to neutralize the interaction of DC-SIGN with ICAM-3/CD50. It should be noted that the DC-SIGN receptor is a tetramer protein composed of four individual domains: the C-terminal carbohydrate recognition domain (CRD), the neck-repeat region, the transmembrane domain, and the N terminal cytoplasmic tail. Due to DC-SIGN’s role in the process of viral infection and its structural complexity, many groups believe that blocking the sugar binding site, ICAM-3 epitope in the carbohydrate recognition domain (CRD) will overall inhibit DC-SIGN and not allow the virus to enter. This assumption is believed to be true because the ICAM-3 epitope is located in the center of the carbohydrate recognition domain where most molecular processes are mediated in the DC-SIGN receptor [
19]. Importantly, the biochemistry of any interactions between DC-SIGN and KSHV glycoproteins has not been established. The fact that our results show increased infection with DC-SIGN neutralization could imply that the neutralizing antibody may be binding to a different epitope on DC-SIGN and that the antibody is not neutralizing the actual interaction of the viral glycoproteins to DC-SIGN. More research is needed to decipher the real interactions between DC-SIGN and KSHV glycoproteins to be able to fully neutralize those specific interactions. Moreover, our results with DC-SIGN neutralization highlight the need to better establish the role of DC-SIGN signaling in our tonsil lymphocyte cultures and its impact on KSHV infection.
Future studies are also needed to determine if the other conserved glycoproteins: gB, gM, gN, ORF4 and gpK8.1 are essential for KSHV entry in tonsil lymphocytes. Through such studies, we will have the opportunity to unravel different entry mechanisms for KSHV in tonsil derived B cells and to determine what viral glycoproteins are essential for entry. In addition, future studies are needed to determine the specific cellular receptors that KSHV uses to enter each susceptible B cell subset and how these receptors change based on inflammatory and metabolic factors that may influence the spread of KSHV within a human host. Previous studies have shown that cellular receptors for KSHV can be modulated by metabolic factors such as high glucose associated with diabetes [
20]. Moreover, KSHV replication has been shown to modify metabolic parameters such as lipogenesis [
21], creating a positive feedback loop between viral spread, persistence and pathogenesis. Understanding the fundamental virology of KSHV entry in the lymphocyte compartment will be essential to rationally designing treatment strategies that limit KSHV spread and pathogenesis within a human host. Moreover, the rational design of vaccines to prevent the person-to-person spread of KSHV, and thereby limit the public health impact of KSHV-associated malignancies, will depend upon our understanding of how KSHV invades the immunological compartment. The results presented herein represent a critical first step in filling this critical gap in our understanding of KSHV virology.



 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}