
Consequence of Histoincompatibility beyond GvH-Reaction in Cytomegalovirus Disease Associated with Allogeneic Hematopoietic Cell Transplantation: Change of Paradigm



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Key Results from Mouse Models of Allogeneic HCT
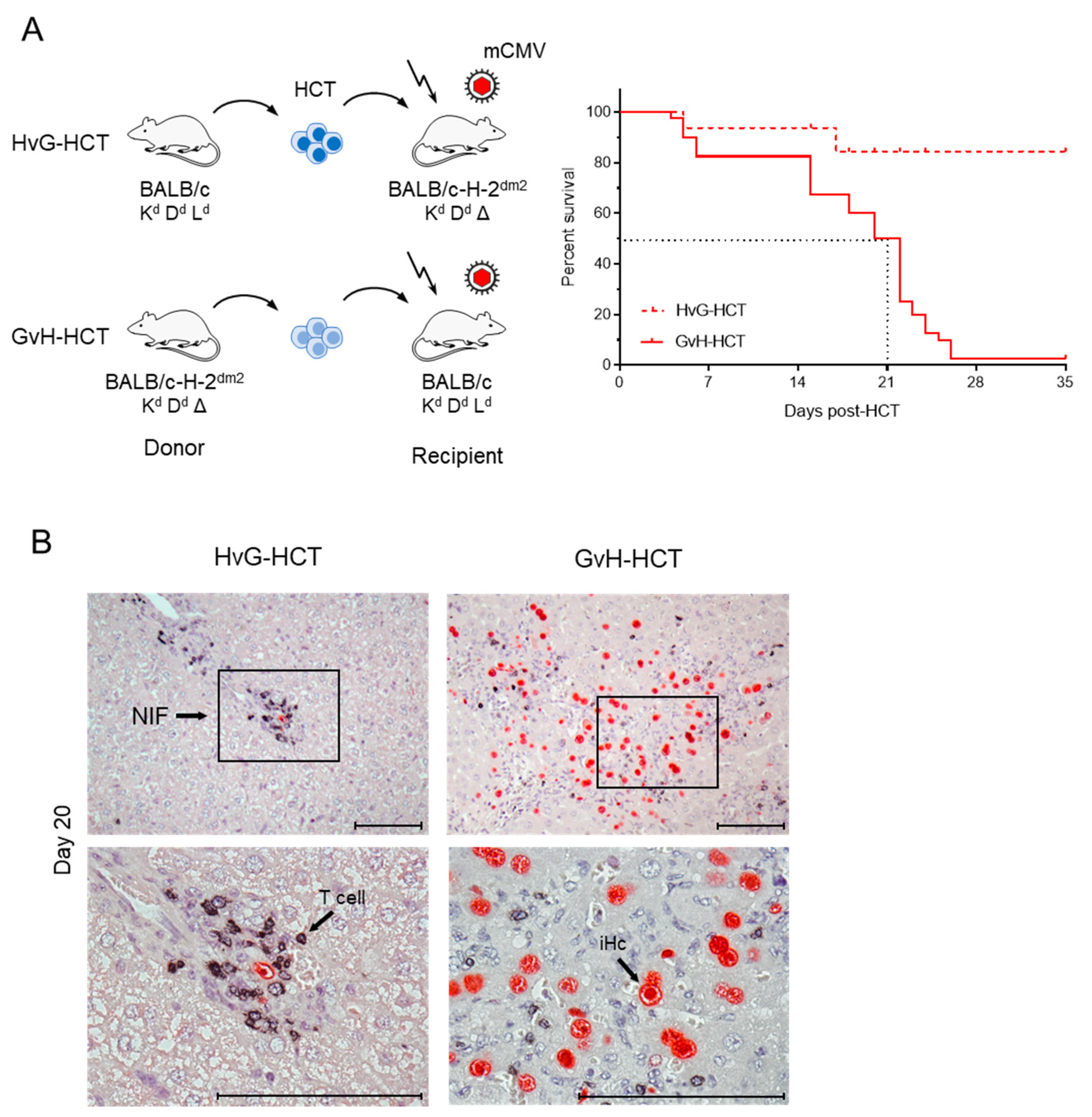
2.1. Lethality from CMV Infection after HCT in Immunogenetic GvH Transplantation Direction
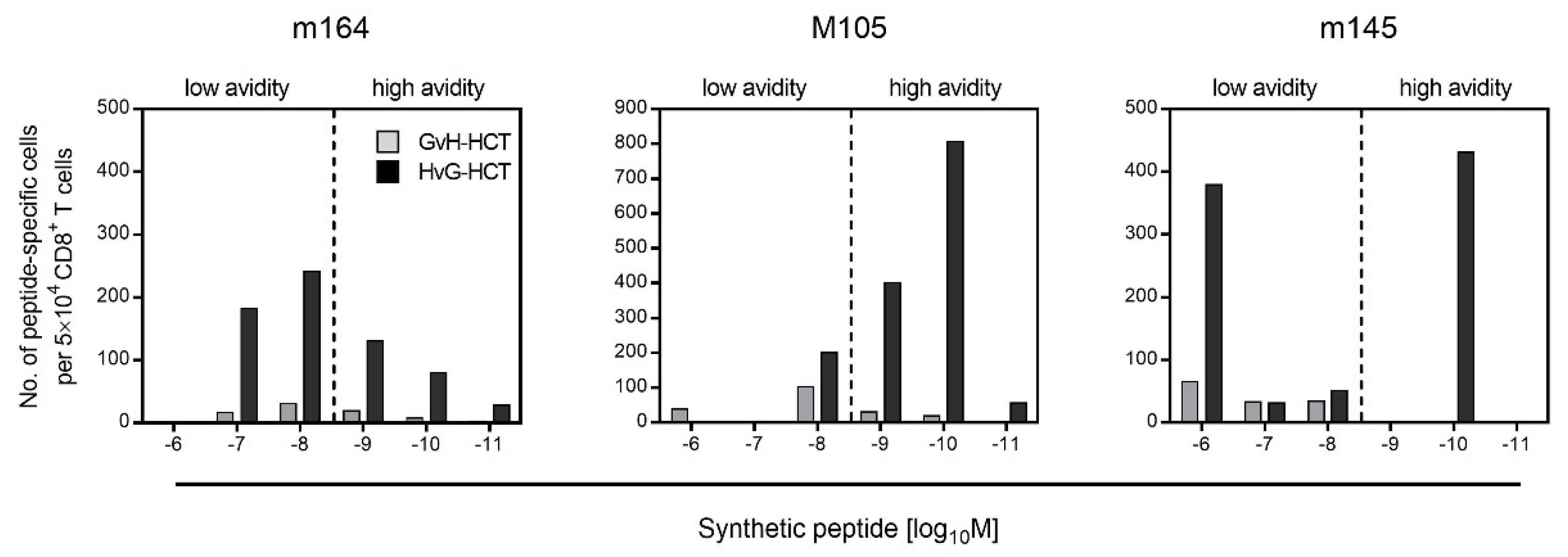
2.2. Failure in the Reconstitution of High Avidity Virus-Specific CD8+ T Cells after GvH-HCT
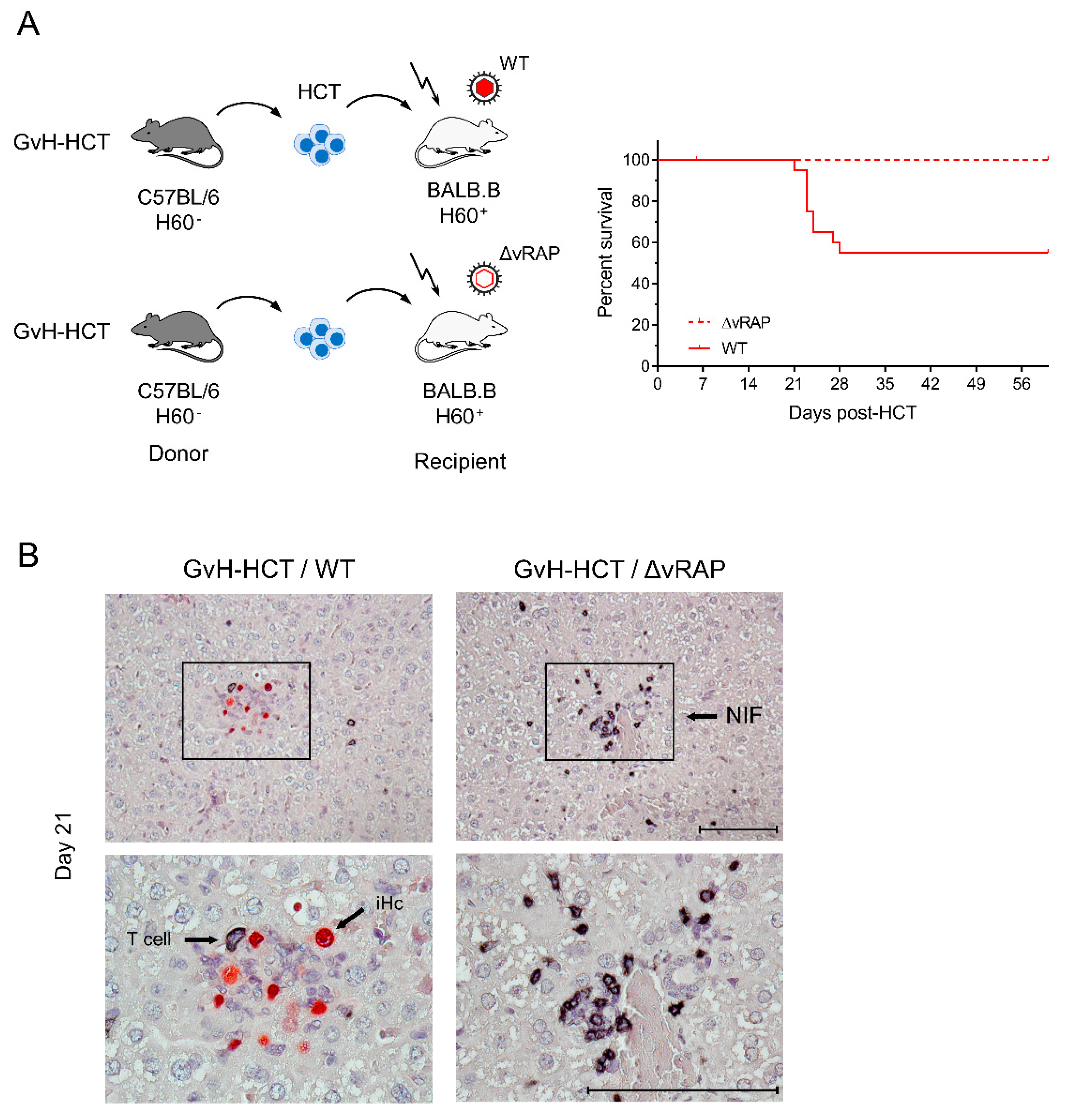
2.3. Enhancement of Antigen Presentation Restores Antiviral Protection after GvH-HCT
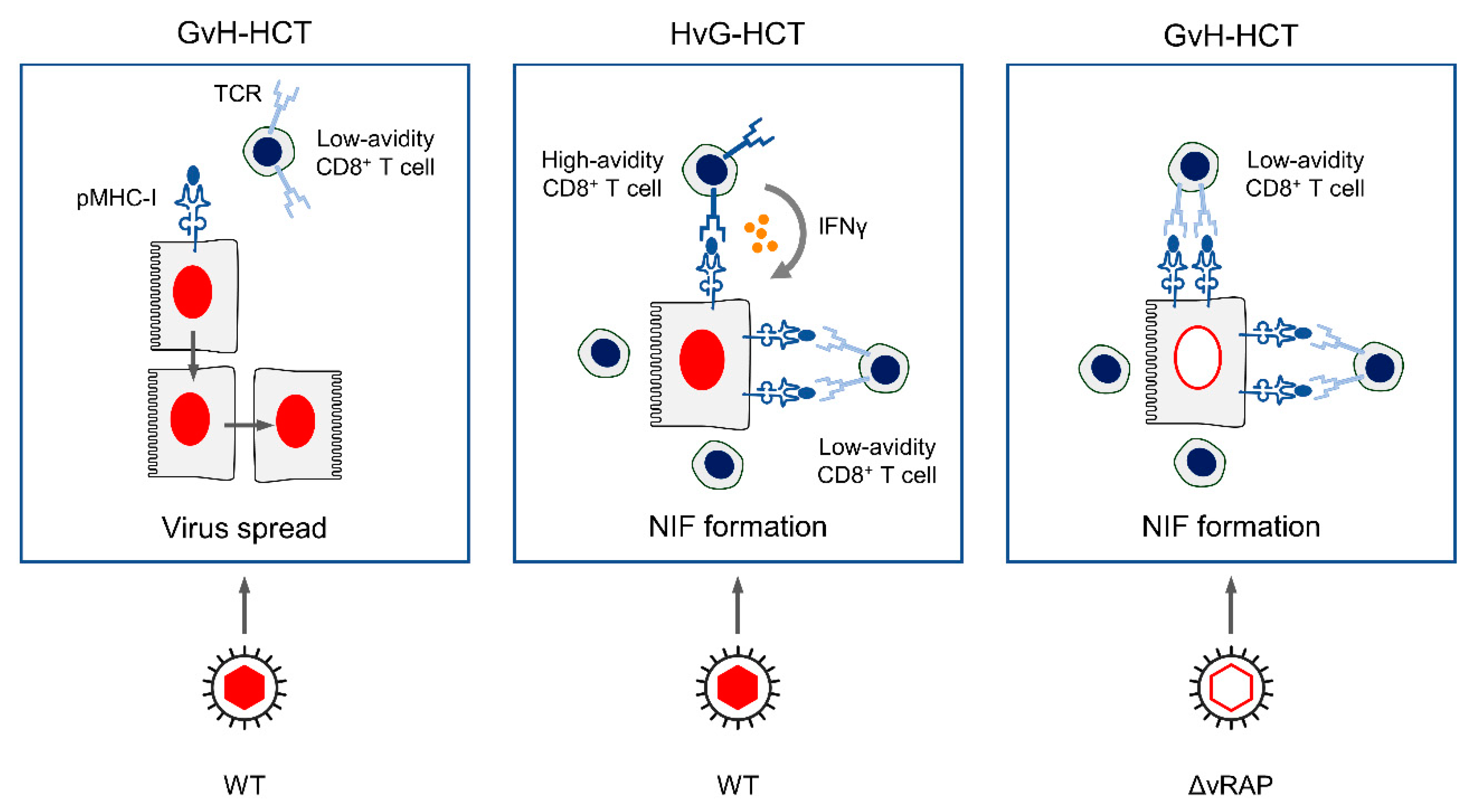
3. Summary
4. Conclusions
5. Lesson Learned for Better Clinical Understanding
Author Contributions
Funding
Conflicts of Interest
References
- Davison, A.J.; Holton, M.; Dolan, A.; Dargan, D.J.; Gatherer, D.; Hayward, G.S. Comparative genomics of primate cytomegaloviruses. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume I, pp. 1–22. [Google Scholar]
- Cannon, M.J.; Grosse, S.D.; Fowler, K.B. The epidemiology and public health impact of congenital cytomegalovirus infection. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume II, pp. 26–48. [Google Scholar]
- Tabata, T.; Petitt, M.; Fang-Hoover, J.; Pereira, L. Survey of cellular immune responses to human cytomegalovirus infection in the microenvironment of the uterine-placental interface. Med. Microbiol. Immunol. 2019, 208, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Britt, W. Manifestations of human cytomegalovirus infection: Proposed mechanisms of acute and chronic disease. Curr. Top. Microbiol. Immunol. 2008, 325, 417–470. [Google Scholar]
- Ho, M. The history of cytomegalovirus and its diseases. Med. Microbiol. Immunol. 2008, 197, 65–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boppana, S.B.; Britt, W.J. Synopsis of clinical aspects of human cytomegalovirus disease. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume I, pp. 1–25. [Google Scholar]
- Adler, S.P.; Nigro, G. Clinical cytomegalovirus research: Congenital infection. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume II, pp. 55–73. [Google Scholar]
- Avery, R.K. Clinical cytomegalovirus research: Thoracic organ transplantation. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume II, pp. 286–300. [Google Scholar]
- Emery, V.C.; Milne, R.S.B.; Griffiths, P.D. Clinical cytomegalovirus research: Liver and kidney transplantation. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume II, pp. 301–311. [Google Scholar]
- Reddehase, M.J.; Lemmermann, N.A.W. Cellular reservoirs of latent cytomegaloviruses. Med. Microbiol. Immunol. 2019, 208, 391–403. [Google Scholar] [CrossRef]
- Riddell, S.R. Pathogenesis of cytomegalovirus pneumonia in immunocompromised hosts. Semin. Respir. Infect. 1995, 10, 199–208. [Google Scholar] [PubMed]
- Seo, S.; Boeckh, M. Clinical cytomegalovirus research: Hematopoietic cell transplantation. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume II, pp. 337–353. [Google Scholar]
- Chemaly, R.F.; Chou, S.; Einsele, H.; Griffiths, P.; Avery, R.; Razonable, R.R.; Mullane, K.M.; Kotton, C.; Lundgren, J.; Komatsu, T.E.; et al. Definitions of resistant and refractory cytomegalovirus infection and disease in transplant recipients for use in clinical trials. Clin. Infect. Dis. 2019, 68, 1420–1426. [Google Scholar] [CrossRef]
- Riddell, S.R.; Watanabe, K.S.; Goodrich, M.; Li, C.R.; Agha, M.E.; Greenberg, P.D. Restoration of viral immunity in immunodeficient humans by the adoptive transfer of T cell clones. Science 1992, 257, 238–241. [Google Scholar] [CrossRef] [PubMed]
- Walter, E.A.; Greenberg, P.D.; Gilbert, M.J.; Finch, R.J.; Watanabe, K.S.; Thomas, D.; Riddell, S.R. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N. Engl. J. Med. 1995, 333, 1038–1044. [Google Scholar] [CrossRef]
- Einsele, H.; Roosnek, E.; Rufer, N.; Sinzger, C.; Riegler, S.; Löffler, J.; Grigoleit, U.; Moris, A.; Rammensee, H.G.; Kanz, L.; et al. Infusion of cytomegalovirus (CMV)-specific T cells for the treatment of CMV infection not responding to antiviral chemotherapy. Blood 2002, 99, 3916–3922. [Google Scholar] [CrossRef] [Green Version]
- Feuchtinger, T.; Opherk, K.; Bethge, W.A.; Topp, M.S.; Schuster, F.R.; Weissinger, E.M.; Mothy, M.; Or, R.; Mashan, M.; Schumm, M.; et al. Adoptive transfer of pp65-specific T cells for the treatment of chemorefractory cytomegalovirus disease or reactivation after haploidentical and matched unrelated stem cell transplantation. Blood 2010, 116, 4360–4367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, L.; Cowan, M.J.; Dunham, K.; Horn, B.; McGuirk, J.; Gilman, A.; Lucas, K.G. Adoptive immunotherapy with CMV-specific cytotoxic T lymphocytes for stem cell transplant patients with refractory CMV infections. J. Immunother. 2012, 35, 293–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odendahl, M.; Grigoleit, G.U.; Bönig, H.; Neuenhahn, M.; Albrecht, J.; Anderl, F.; Germeroth, L.; Schmitz, M.; Bornhäuser, M.; Einsele, H.; et al. Clinical-scale isolation of ’minimally manipulated’ cytomegalovirus-specific donor lymphocytes for the treatment of refractory cytomegalovirus disease. Cytotherapy 2014, 16, 1245–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, X.Y.; Zhao, X.Y.; Chang, Y.J.; Liu, J.; Xu, L.P.; Wang, Y.; Zhang, X.H.; Han, W.; Chen, Y.H.; Huang, X.J. Cytomegalovirus-specific T-cell transfer for refractory cytomegalovirus infection after haploidentical stem cell transplantation: The quantitative and qualitative immune recovery for cytomegalovirus. J. Infect. Dis. 2017, 216, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Kaeuferle, T.; Krauss, R.; Blaeschke, F.; Willier, S.; Feuchtinger, T. Strategies of adoptive T -cell transfer to treat refractory viral infections post allogeneic stem cell transplantation. J. Hematol. Oncol. 2019, 12, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rappeport, J.; Mihm, M.; Reinherz, E.; Lopansri, S.; Parkman, R. Acute graft-versus-host disease in recipients of bone-marrow transplants from identical twin donors. Lancet 1979, 2, 717–720. [Google Scholar] [CrossRef]
- Applebaum, F.R.; Meyers, J.D.; Fefer, A.; Fluornoy, N.; Cheever, M.A.; Greenberg, P.D.; Hackman, R.; Thomas, E.D. Nonbacterial nonfungal pneumonia following marrow transplantation in 100 identical twins. Transplantation 1982, 33, 265–268. [Google Scholar]
- Wingard, J.R.; Chen, D.Y.; Burns, W.H.; Fuller, D.J.; Braine, H.G.; Yeager, A.M.; Kaiser, H.; Burke, P.J.; Graham, M.L.; Santos, G.W.; et al. Cytomegalovirus infection after autologous bone marrow transplantation with comparison to infection after allogeneic bone marrow transplantation. Blood 1988, 71, 1432–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyers, J.D.; Flournoy, N.; Thomas, E.D. Nonbacterial pneumonia after allogeneic marrow transplantation: A review of ten years’ experience. Rev. Infect. Dis. 1982, 4, 1119–1132. [Google Scholar] [CrossRef]
- Bleakley, M.D.; Brown, M.; Riddell, S. Human CD8+ minor histocompatibility antigen specific cytotoxic T lymphocyte clones can be generated by primary in vitro stimulation of naїve T cells with dendritic cells from HLA identical siblings. Blood 2004, 104, 2116. [Google Scholar] [CrossRef]
- Stern, L.; Withers, B.; Avdic, S.; Gottlieb, D.; Abendroth, A.; Blyth, E.; Slobedman, B. Human cytomegalovirus latency and reactivation in allogeneic hematopoietic stem cell transplant recipients. Front. Microbiol. 2019, 10, 1186. [Google Scholar] [CrossRef] [Green Version]
- Hartwig, U.F.; Nonn, M.; Khan, S.; Link, I.; Huber, C.; Herr, W. Depletion of alloreactive donor T lymphocytes by CD95-mediated activation-induced cell death retains antileukemic, antiviral, and immunoregulatory T cell immunity. Biol. Blood Marrow Transplant. 2008, 14, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.; Alawieh, A.; Bastian, D.; Kuril, S.; Dai, M.; Daenthanasanmak, A.; Zhang, M.; Iamsawat, S.; Schutt, S.D.; Wu, Y.; et al. Targeting the complement alternative pathway permits graft versus leukemia activity while preventing graft versus host disease. Clin. Cancer Res. 2020, 26, 3481–3490. [Google Scholar] [CrossRef] [Green Version]
- Deeg, H.J. Chimerism, the microenvironment and control of leukemia. Front. Immunol. 2021, 12, 652105. [Google Scholar] [CrossRef]
- Emery, V.C. Relative importance of cytomegalovirus load as a risk factor for cytomegalovirus disease in the immunocompromised host. Monogr. Virol. 1998, 21, 288–301. [Google Scholar]
- Sykes, M. Hematopoietic cell transplantation for tolerance induction: Animal models to clinical trials. Transplantation 2009, 87, 309–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddehase, M.J.; Lemmermann, N.A.W. Mouse model of cytomegalovirus disease and immunotherapy in the immunocompromised host: Predictions for medical translation that survived the “test of time”. Viruses 2018, 10, 693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddehase, M.J.; Weiland, F.; Münch, K.; Jonjic, S.; Lüske, A.; Koszinowski, U.H. Interstitial murine cytomegalovirus pneumonia after irradiation: Characterization of cells that limit viral replication during established infection of the lungs. J. Virol. 1985, 55, 264–273. [Google Scholar] [CrossRef] [Green Version]
- Reddehase, M.J.; Mutter, W.; Koszinowski, U.H. In vivo application of recombinant interleukin 2 in the immunotherapy of established cytomegalovirus infection. J. Exp. Med. 1987, 165, 650–656. [Google Scholar] [CrossRef] [Green Version]
- Ebert, S.; Podlech, J.; Gillert-Marien, D.; Gergely, K.M.; Büttner, J.K.; Fink, A.; Freitag, K.; Thomas, D.; Reddehase, M.J.; Holtappels, R. Parameters determining the efficacy of adoptive CD8 T-cell therapy of cytomegalovirus infection. Med. Microbiol. Immunol. 2012, 201, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Holtappels, R.; Ebert, S.; Podlech, J.; Fink, A.; Böhm, V.; Lemmermann, N.A.W.; Freitag, K.; Renzaho, A.; Thomas, D.; Reddehase, M.J. Murine model for cytoimmunotherapy of CMV disease after haematopoietic cell transplantation. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume II, pp. 352–379. [Google Scholar]
- Reddehase, M.J. Mutual interference between cytomegalovirus and reconstitution of protective immunity after hematopoietic cell transplantation. Front. Immunol. 2016, 7, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adler, S.P.; Reddehase, M.J. Pediatric roots of cytomegalovirus recurrence and memory inflation in the elderly. Med. Microbiol. Immunol. 2019, 208, 323–328. [Google Scholar] [CrossRef]
- Adler, B.; Sinzger, C. Cytomegalovirus interstrain variance in cell type tropism. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume I, pp. 297–321. [Google Scholar]
- Wilkinson, G.W.; Davison, A.J.; Tomasec, P.; Fielding, C.A.; Aicheler, R.; Murrell, I.; Seirafian, S.; Wang, E.C.; Weekes, M.; Lehner, P.J.; et al. Human cytomegalovirus: Taking the strain. Med. Microbiol. Immunol. 2015, 204, 273–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtappels, R.; Schader, S.I.; Oettel, O.; Podlech, J.; Seckert, C.K.; Reddehase, M.J.; Lemmermann, N.A.W. Insufficient antigen presentation due to viral immune evasion explains lethal cytomegalovirus organ disease after allogeneic hematopoietic cell transplantation. Front. Cell. Infect. Microbiol. 2020, 10, 157. [Google Scholar] [CrossRef] [Green Version]
- Gezinir, E.; Podlech, J.; Gergely, K.M.; Becker, S.; Reddehase, M.J.; Lemmermann, N.A.W. Enhancement of antigen presentation by deletion of viral immune evasion genes prevents lethal cytomegalovirus disease in minor histocompatibility antigen-mismatched hematopoietic cell transplantation. Front. Cell. Infect. Microbiol. 2020, 10, 279. [Google Scholar] [CrossRef] [PubMed]
- Holtappels, R.; Freitag, K.; Renzaho, A.; Becker, S.; Lemmermann, N.A.W.; Reddehase, M.J. Revisiting CD8 T-cell ‘memory inflation’: New insights with implications for cytomegaloviruses as vaccine vectors. Vaccines 2020, 8, 402. [Google Scholar] [CrossRef]
- Roopenian, D.; Choi, E.Y.; Brown, A. The immunogenomics of minor histocompatibility antigens. Immunol. Rev. 2002, 190, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.; Krmpotic, A.; Ruzsics, Z.; Bubic, I.; Lenac, T.; Halenius, A.; Loewendorf, A.; Messerle, M.; Hengel, H.; Jonjic, S.; et al. Selective down-regulation of the NKG2D ligand H60 by mouse cytomegalovirus m155 glycoprotein. J. Virol. 2005, 79, 2920–2930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, K.J.; Bushell, A.; Hester, J. Regulatory immune cells in transplantation. Nat. Rev. Immunol. 2012, 12, 417–430. [Google Scholar] [CrossRef]
- Fink, A.; Lemmermann, N.A.; Gillert-Marien, D.; Thomas, D.; Freitag, K.; Böhm, V.; Wilhelmi, V.; Reifenberg, K.; Reddehase, M.J.; Holtappels, R. Antigen presentation under the influence of ’immune evasion’ proteins and its modulation by interferon-gamma: Implications for immunotherapy of cytomegalovirus infection with antiviral CD8 T cells. Med. Microbiol. Immunol. 2012, 201, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Gratama, J.W.; van Esser, J.W.; Lamers, C.H.; Tournay, C.; Löwenberg, B.; Bolhuis, R.L.; Cornelissen, J.J. Tetramer-based quantification of cytomegalovirus (CMV)-specific CD8+ T lymphocytes in T-cell-depleted stem cell grafts and after transplantation may identify patients at risk for progressive CMV infection. Blood 2001, 98, 1358–1364. [Google Scholar] [CrossRef] [Green Version]
- Cwynarski, K.; Ainsworth, J.; Cobbold, M.; Wagner, S.; Mahendra, P.; Apperley, J.; Goldman, J.; Craddock, C.; Moss, P.A. Direct visualization of cytomegalovirus-specific T-cell reconstitution after allogeneic stem cell transplantation. Blood 2001, 97, 1232–1240. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reddehase, M.J.; Holtappels, R.; Lemmermann, N.A.W. Consequence of Histoincompatibility beyond GvH-Reaction in Cytomegalovirus Disease Associated with Allogeneic Hematopoietic Cell Transplantation: Change of Paradigm. Viruses 2021, 13, 1530. https://doi.org/10.3390/v13081530
Reddehase MJ, Holtappels R, Lemmermann NAW. Consequence of Histoincompatibility beyond GvH-Reaction in Cytomegalovirus Disease Associated with Allogeneic Hematopoietic Cell Transplantation: Change of Paradigm. Viruses. 2021; 13(8):1530. https://doi.org/10.3390/v13081530
Chicago/Turabian StyleReddehase, Matthias J., Rafaela Holtappels, and Niels A. W. Lemmermann. 2021. "Consequence of Histoincompatibility beyond GvH-Reaction in Cytomegalovirus Disease Associated with Allogeneic Hematopoietic Cell Transplantation: Change of Paradigm" Viruses 13, no. 8: 1530. https://doi.org/10.3390/v13081530
APA StyleReddehase, M. J., Holtappels, R., & Lemmermann, N. A. W. (2021). Consequence of Histoincompatibility beyond GvH-Reaction in Cytomegalovirus Disease Associated with Allogeneic Hematopoietic Cell Transplantation: Change of Paradigm. Viruses, 13(8), 1530. https://doi.org/10.3390/v13081530