
HTS-Based Diagnostics of Sugarcane Viruses: Seasonal Variation and Its Implications for Accurate Detection

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Seasonal Sample Collection
2.2. RNA Extraction, Library Preparation and Sequencing
2.3. HTS Data Analysis: Normalization, Mapping, and De Novo Assembly
2.4. Normalized Read Count Calculation and Read Sub-Sampling
2.5. Quantitative RT-PCR of Targeted Viruses
3. Results
3.1. Identification of DNA and RNA Viruses: Comparison between HTS and Conventional Indexing Methods
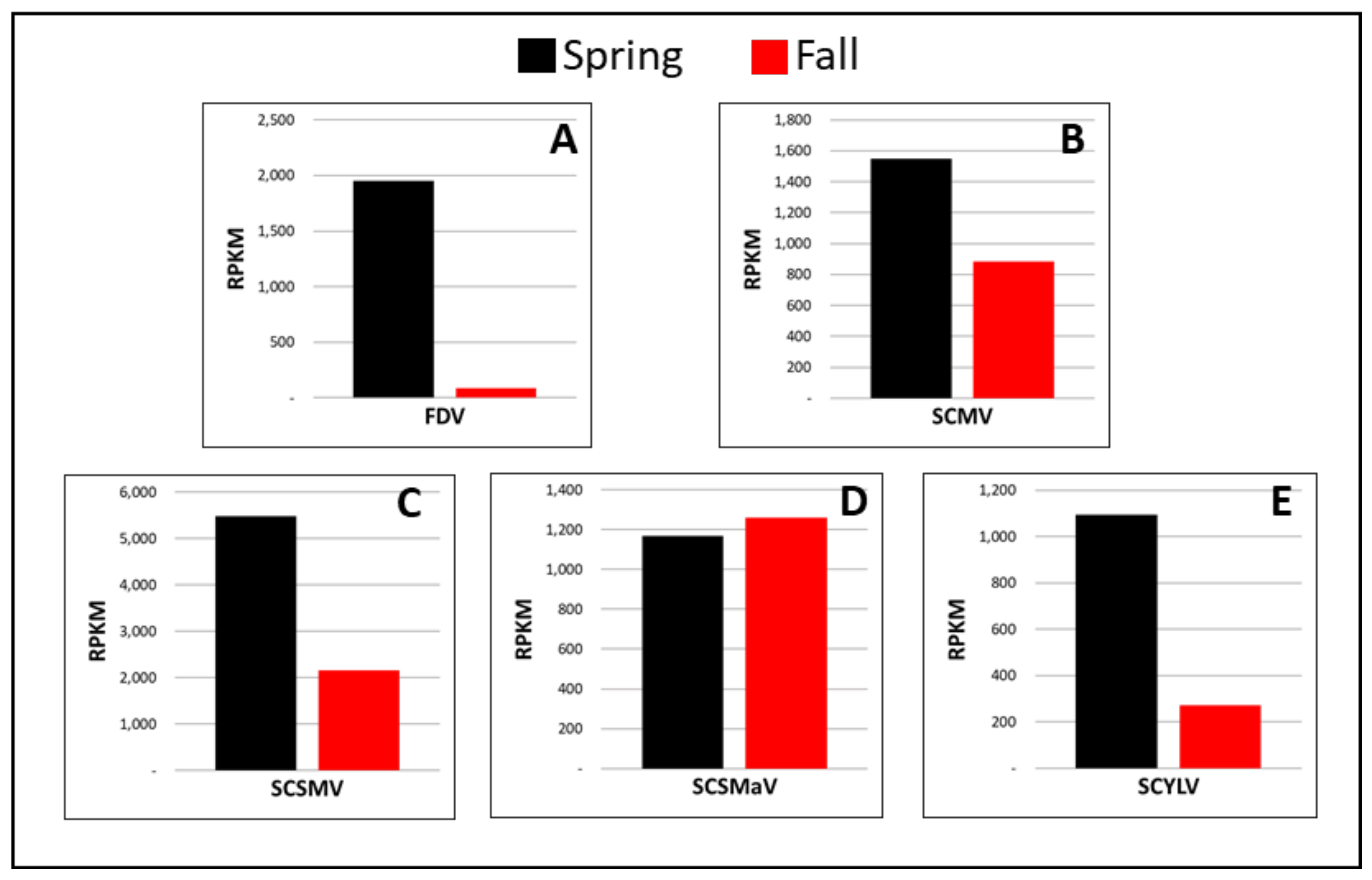
3.2. Comparison of Viral RPKM-Normalized Reads between Seasons
3.2.1. RNA Viruses
3.2.2. DNA Viruses
3.3. Validation of Seasonal Viral Titer Change Using RT-qPCR
3.4. Reproducibility: Comparison of SCYLV RPKM Variation
3.5. Seasonal Viral Identification: Comparison of Sequencing Depth
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oerke, E.C. Crop losses to pests. J. Agric. Sci. 2006, 144, 31–43. [Google Scholar] [CrossRef]
- Savary, S.; Willocquet, L.; Pethybridge, S.J.; Esker, P.; McRoberts, N.; Nelson, A. The global burden of pathogens and pests on major food crops. Nat. Ecol. Evol. 2019, 3, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Sastry, K.S.; Zitter, T.A. Management of Virus and Viroid Disease of Crops in the Tropics. In Plant Virus and Viroid Diseases in the Tropics; Springer: Dordrecht, The Netherlands, 2014; pp. 149–480. [Google Scholar]
- Rybicki, E.P. A Top Ten list for economically important plant viruses. Arch. Virol. 2015, 160, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Byrne, P.F.; Volk, G.M.; Gardner, C.; Gore, M.A.; Simon, P.W.; Smith, S. Sustaining the future of plant breeding: The critical role of the USDA-ARS national plant germplasm system. Crop Sci. 2018, 58, 451–468. [Google Scholar] [CrossRef] [Green Version]
- Savary, S.; Teng, P.S.; Willocquet, L.; Nutter, F.W. Quantification and modeling of crop losses: A review of purposes. Annu. Rev. Phytopathol. 2006, 44, 89–112. [Google Scholar] [CrossRef] [PubMed]
- Levy, L.; Damsteegt, V.; Welliver, R. First Report of Plum pox virus (Sharka Disease) in Prunus persica in the United States. Plant Dis. 2000, 84, 202. [Google Scholar] [CrossRef]
- Cambra, M.; Capote, N.; Myrta, A.; Llácer, G. Plum pox virus and the estimated costs associated with sharka disease. EPPO Bull. 2006, 36, 202–204. [Google Scholar] [CrossRef]
- Barba, M.; Hadidi, A.; Candresse, T.; Cambra, M. Plum pox virus. In Virus and Virus-Like Diseases of Pome and Stone Fruits; Hadidi, A., Barba, M., Candress, T., Jelkmann, W., Eds.; American Phytopathological Society: St. Paul, MN, USA, 2011; pp. 185–197. [Google Scholar]
- USDA Declares United States Free from Plum Pox Virus. Available online: https://www.usda.gov/media/press-releases/2019/10/17/usda-declares-united-states-free-plum-pox-virus (accessed on 28 July 2021).
- Carvajal-Yepes, M.; Cardwell, K.; Nelson, A.; Garrett, K.A.; Giovani, B.; Saunders, D.G.O.; Kamoun, S.; Legg, J.P.; Verdier, V.; Lessel, J.; et al. A global surveillance system for crop diseases. Science 2019, 364, 1237–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strange, R.N.; Scott, P.R. Plant disease: A threat to global food security. Annu. Rev. Phytopathol. 2005, 43, 83–116. [Google Scholar] [CrossRef] [PubMed]
- Animal and Plant Health Inspection Service; US. Department of Agriculture. Plant Quarantine Programs Managed by APHIS-PPQ. Available online: https://www.aphis.usda.gov/aphis/ourfocus/planthealth/import-information/permits/plants-and-plant-products-permits/prohibited/Importation-of-Plant-Parts-for-Propagation/ (accessed on 28 July 2021).
- Grof, C.P.L.; Campbell, J.A. Sugarcane sucrose metabolism: Scope for molecular manipulation. Aust. J. Plant Physiol. 2001, 28, 1–12. [Google Scholar] [CrossRef]
- Dal-Bianco, M.; Carneiro, M.S.; Hotta, C.T.; Chapola, R.G.; Hoffmann, H.P.; Garcia, A.A.F.; Souza, G.M. Sugarcane improvement: How far can we go? Curr. Opin. Biotechnol. 2012, 23, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Huang, N.; Wu, Q.; Su, Y.; Peng, Q.; Ahmed, W.; Gao, S.; Su, W.; Que, Y.; Xu, L. Transcriptional Insights into the Sugarcane-Sorghum mosaic virus Interaction. Trop. Plant Biol. 2018, 11, 163–176. [Google Scholar] [CrossRef]
- Viswanathan, R.; Parameswari, B.; Nithya, K. Molecular Characterization of Sugarcane Viruses and Their Diagnostics. In Crop Improvement through Microbial Biotechnology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 175–193. [Google Scholar]
- Olmos, A.; Boonham, N.; Candresse, T.; Gentit, P.; Giovani, B.; Kutnjak, D.; Liefting, L.; Maree, H.J.; Minafra, A.; Moreira, A.; et al. High-throughput sequencing technologies for plant pest diagnosis: Challenges and opportunities. EPPO Bull. 2018, 48, 219–224. [Google Scholar] [CrossRef] [Green Version]
- Villamor, D.E.V.; Ho, T.; Al Rwahnih, M.; Martin, R.R.; Tzanetakis, I.E. High throughput sequencing for plant virus detection and discovery. Phytopathology 2019, 109, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Al Rwahnih, M.; Daubert, S.; Golino, D.; Rowhani, A. Deep sequencing analysis of RNAs from a grapevine showing Syrah decline symptoms reveals a multiple virus infection that includes a novel virus. Virology 2009, 387, 395–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Candresse, T.; Filloux, D.; Muhire, B.; Julian, C.; Galzi, S.; Fort, G.; Bernardo, P.; Daugrois, J.H.; Fernandez, E.; Martin, D.P.; et al. Appearances can be deceptive: Revealing a hidden viral infection with deep sequencing in a plant quarantine context. PLoS ONE 2014, 9, e0102945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiumenti, M.; Torchetti, E.M.; Di Serio, F.; Minafra, A. Identification and characterization of a viroid resembling apple dimple fruit viroid in fig (Ficus carica L.) by next generation sequencing of small RNAs. Virus Res. 2014, 188, 54–59. [Google Scholar] [CrossRef]
- Bag, S.; Al Rwahnih, M.; Li, A.; Gonzalez, A.; Rowhani, A.; Uyemoto, J.K.; Sudarshana, M.R. Detection of a new luteovirus in imported nectarine trees: A case study to propose adoption of metagenomics in post-entry quarantine. Phytopathology 2015, 105, 840–846. [Google Scholar] [CrossRef] [Green Version]
- Katsiani, A.; Maliogka, V.I.; Katis, N.; Svanella-Dumas, L.; Olmos, A.; Ruiz-García, A.B.; Marais, A.; Faure, C.; Theil, S.; Lotos, L.; et al. High-throughput sequencing reveals further diversity of little cherry virus 1 with implications for diagnostics. Viruses 2018, 10, 385. [Google Scholar] [CrossRef] [Green Version]
- Bejerman, N.; Debat, H.; Dietzgen, R.G. The Plant Negative-Sense RNA Virosphere: Virus Discovery through New Eyes. Front. Microbiol. 2020, 11, 588427. [Google Scholar] [CrossRef]
- Beris, D.; Ioanna, M.; Vassilakos, N.; Theologidis, I.; Rampou, A.; Kektsidou, O.; Massart, S.; Varveri, C. Association of citrus virus A to citrus impietratura disease symptoms. Phytopathology 2021, 1–44. [Google Scholar] [CrossRef]
- Adams, I.; Fox, A. Diagnosis of Plant Viruses Using Nest-Generation Sequencing and Mategenomic Analysis. In Current Research Topics in Plant Virology; Wang, A., Zhou, X., Eds.; Springer: Cham, Switzerland, 2016; pp. 323–335. [Google Scholar]
- Al Rwahnih, M.; Daubert, S.; Golino, D.; Islas, C.; Rowhani, A. Comparison of next-generation sequencing versus biological indexing for the optimal detection of viral pathogens in grapevine. Phytopathology 2015, 105, 758–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bester, R.; Cook, G.; Breytenbach, J.H.J.; Steyn, C.; De Bruyn, R.; Maree, H.J. Towards the validation of high-throughput sequencing (HTS) for routine plant virus diagnostics: Measurement of variation linked to HTS detection of citrus viruses and viroids. Virol. J. 2021, 18, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Maree, H.J.; Fox, A.; Al Rwahnih, M.; Boonham, N.; Candresse, T. Application of hts for routine plant virus diagnostics: State of the art and challenges. Front. Plant Sci. 2018, 9, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Kinoti, W.M.; Nancarrow, N.; Dann, A.; Rodoni, B.C.; Constable, F.E. Updating the quarantine status of prunus infecting viruses in Australia. Viruses 2020, 12, 246. [Google Scholar] [CrossRef] [Green Version]
- Rott, M.; Xiang, Y.; Boyes, I.; Belton, M.; Saeed, H.; Kesanakurti, P.; Hayes, S.; Lawrence, T.; Birch, C.; Bhagwat, B.; et al. Application of next generation sequencing for diagnostic testing of tree fruit viruses and viroids. Plant Dis. 2017, 101, 1489–1499. [Google Scholar] [CrossRef] [Green Version]
- Honjo, M.N.; Emura, N.; Kawagoe, T.; Sugisaka, J.; Kamitani, M.; Nagano, A.J.; Kudoh, H. Seasonality of interactions between a plant virus and its host during persistent infection in a natural environment. ISME J. 2020, 14, 506–518. [Google Scholar] [CrossRef] [Green Version]
- Roden, L.C.; Ingle, R.A. Lights, rhythms, infection: The role of light and the circadian clock in determining the outcome of plant-pathogen interactions. Plant Cell 2009, 21, 2546–2552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szittya, G.; Silhavy, D.; Molnár, A.; Havelda, Z.; Lovas, Á.; Lakatos, L.; Bánfalvi, Z.; Burgyán, J. Low temperature inhibits RNA silencing-mediated defence by the control of siRNA generation. EMBO J. 2003, 22, 633–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chellappan, P.; Vanitharani, R.; Ogbe, F.; Fauquet, C.M. Effect of temperature on geminivirus-induced RNA silencing in plants. Plant Physiol. 2005, 138, 1828–1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szittya, G.; Burgyán, J. RNA Interference-Mediated Intrinsic Antiviral Immunity in Plants. In Intrinsic Immunity; Cullen, B.R., Ed.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 153–181. [Google Scholar]
- Jones, R.W.; Jackson, A.O.; Morris, T.J. Defective-interfering RNAs and elevated temperatures inhibit replication of tomato bushy stunt virus in inoculated protoplasts. Virology 1990, 176, 539–545. [Google Scholar] [CrossRef]
- Ohsato, S.; Miyanishi, M.; Shirako, Y. The optimal temperature for RNA replication in cells infected by Soil-borne wheat mosaic virus is 17 °C. J. Gen. Virol. 2003, 84, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.N.; Choi, K.S.; Ahn, J.J.; Joa, J.H.; Do, K.S.; Park, K.S. Effects of temperature on systemic infection and symptom expression of turnip mosaic virus in chinese cabbage (Brassica campestris). Plant Pathol. J. 2015, 31, 363–370. [Google Scholar] [CrossRef] [Green Version]
- Chung, B.N.; Canto, T.; Tenllado, F.; Choi, K.S.; Joa, J.H.; Ahn, J.J.; Kim, C.H.; Do, K.S. The effects of high temperature on infection by Potato virus Y, potato virus A, and Potato leafroll virus. Plant Pathol. J. 2016, 32, 321–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Kankainen, M.; Parsons, A.; Kallioniemi, O.; Mattila, P.; Heckman, C.A. The impact of RNA sequence library construction protocols on transcriptomic profiling of leukemia. BMC Genom. 2017, 18, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Bal, A.; Pichon, M.; Picard, C.; Casalegno, J.S.; Valette, M.; Schuffenecker, I.; Billard, L.; Vallet, S.; Vilchez, G.; Cheynet, V.; et al. Quality control implementation for universal characterization of DNA and RNA viruses in clinical respiratory samples using single metagenomic next-generation sequencing workflow. bioRxiv 2018, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hily, J.M.; Candresse, T.; Garcia, S.; Vigne, E.; Tannière, M.; Komar, V.; Barnabé, G.; Alliaume, A.; Gilg, S.; Hommay, G.; et al. High-throughput sequencing and the viromic study of grapevine leaves: From the detection of grapevine-infecting viruses to the description of a new environmental Tymovirales member. Front. Microbiol. 2018, 9, 1782. [Google Scholar] [CrossRef]
- Vigne, E.; Garcia, S.; Komar, V.; Lemaire, O.; Hily, J.M. Comparison of serological and molecular methods with high-throughput sequencing for the detection and quantification of grapevine fanleaf virus in vineyard samples. Front. Microbiol. 2018, 9, 2726. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Visser, M.; Bester, R.; Burger, J.T.; Maree, H.J. Next-generation sequencing for virus detection: Covering all the bases. Virol. J. 2016, 13, 4–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Marais, A.; Lefebvre, M.; Theil, S.; Svanella-Dumas, L.; Faure, C.; Candresse, T. Phytovirome Analysis of Wild Plant Populations: Comparison of Double-Stranded RNA and Virion-Associated Nucleic Acid Metagenomic Approaches. J. Virol. 2019, 94, e01462-19. [Google Scholar] [CrossRef] [PubMed]
- Pecman, A.; Kutnjak, D.; Gutiérrez-Aguirre, I.; Adams, I.; Fox, A.; Boonham, N.; Ravnikar, M. Next generation sequencing for detection and discovery of plant viruses and viroids: Comparison of two approaches. Front. Microbiol. 2017, 8, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massart, S.; Chiumenti, M.; De Jonghe, K.; Glover, R.; Haegeman, A.; Koloniuk, I.; Komínek, P.; Kreuze, J.; Kutnjak, D.; Lotos, L.; et al. Virus detection by high-throughput sequencing of small RNAs: Large-scale performance testing of sequence analysis strategies. Phytopathology 2019, 109, 488–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maliogka, V.I.; Minafra, A.; Saldarelli, P.; Ruiz-García, A.B.; Glasa, M.; Katis, N.; Olmos, A. Recent advances on detection and characterization of fruit tree viruses using high-throughput sequencing technologies. Viruses 2018, 10, 436. [Google Scholar] [CrossRef] [Green Version]
- Süss, J.; Schrader, C.; Abel, U.; Voigt, W.P.; Schosser, R. Annual and seasonal variation of tick-borne encephalitis virus (TBEV) prevalence in ticks in selected hot spot areas in Germany using a nRT-PCR: Results from 1997 and 1998. Zent. Bakteriol. 1999, 289, 564–578. [Google Scholar] [CrossRef]
- Withyachumnarnkul, B.; Boonsaeng, V.; Chomsoong, R.; Flegel, T.W.; Muangsin, S.; Nash, G.L. Seasonal variation in white spot syndrome virus-positive samples in broodstock and post-larvae of Penaeus monodon in Thailand. Dis. Aquat. Organ. 2003, 53, 167–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dijk, J.G.B.; Hoye, B.J.; Verhagen, J.H.; Nolet, B.A.; Fouchier, R.A.M.; Klaassen, M. Juveniles and migrants as drivers for seasonal epizootics of avian influenza virus. J. Anim. Ecol. 2014, 83, 266–275. [Google Scholar] [CrossRef] [Green Version]
- Agha, S.B.; Tchouassi, D.P.; Bastos, A.D.S.; Sang, R. Dengue and yellow fever virus vectors: Seasonal abundance, diversity and resting preferences in three Kenyan cities. Parasites Vectors 2017, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Llamas-Llamas, M.E.; Zavaleta-Mejia, E.; Gonzalez-Hernandez, V.A.; Cervantes-Diaz, L.; Santizo-Rincon, J.A.; Ochoa-Martinez, D.L. Effect of temperature on symptom expression and accumulation of tomato spotted wilt virus in different host species. Plant Pathol. 1998, 47, 341–347. [Google Scholar] [CrossRef]
- Hull, R. Replication of Plant Viruses. In Plant Virology; Hull, R., Ed.; Elsevier: London, UK, 2013; pp. 341–422. [Google Scholar]
- Li, Z.; Nagy, P.D. Diverse roles of host RNA binding proteins in RNA virus replication. RNA Biol. 2011, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fondong, V.N. Geminivirus protein structure and function. Mol. Plant Pathol. 2013, 14. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus Species | Viral Family | Genome |
|---|---|---|
| Fiji disease virus (FDV) | Reoviridae | dsRNA |
| Sugarcane mosaic virus (SCMV) | Potyviridae | +ssRNA |
| Sugarcane streak mosaic virus (SCSMV) | Potyviridae | +ssRNA |
| Sugarcane striate mosaic associated virus (SCSMaV) | Betaflexiviridae | +ssRNA |
| Sugarcane yellow leaf virus (SCYLV) | Luteoviridae | +ssRNA |
| Sugarcane streak Egypt virus (SCSEV) | Geminiviridae | ssDNA |
| Sugarcane white streak virus (SCWSV) | Geminiviridae | ssDNA |
| Sample | Origin | Season | Virus Identified 1 | NCBI Accession | Viral Genome (bp) |
|---|---|---|---|---|---|
| P1 | South Africa | Spring | SCYLV-BRA | AF157029 | 5899 |
| SCMV | JX237862 | 9571 | |||
| Fall | SCYLV-BRA | AF157029 | 5899 | ||
| SCMV | JX237862 | 9571 | |||
| P2 | Papua New Guinea | Spring | SCYLV-CHN | GU190159 | 5879 |
| Fall | SCYLV-CHN | GU190159 | 5879 | ||
| P3 | Australia | Spring | SCSMaV | NC_003870 | 8146 |
| Fall | SCSMaV | NC_003870 | 8146 | ||
| P4 | Australia | Spring | FDV | NC_007159 | 4532 |
| Fall | FDV | NC_007159 | 4532 | ||
| P5 | South Africa | Spring | SCSEV | NC_001868 | 2706 |
| SCWSV | NC_023989 | 2830 | |||
| Fall | SCSEV | NC_001868 | 2706 | ||
| SCWSV | NC_023989 | 2830 | |||
| P6 | USA | Spring | SCYLV-CHN | GU190159 | 5879 |
| Fall | SCYLV-CHN | GU190159 | 5879 | ||
| P7 | Pakistan | Spring | SCSMV | NC_014037 | 9782 |
| Fall | SCSMV | NC_014037 | 9782 | ||
| P8 | Guatemala | Spring | SCYLV-BRA | AF157029 | 5899 |
| Fall | SCYLV-BRA | AF157029 | 5899 | ||
| P9 | Guatemala | Spring | SCYLV-BRA | AF157029 | 5899 |
| Fall | SCYLV-BRA | AF157029 | 5899 |
| Sample | Season | Total Number Reads 1 | Virus Identified 2 | No. Viral Reads 3 | RPKM 4 | % Viral Reads 5 | % Viral Genome Coverage | % Identity 6 |
|---|---|---|---|---|---|---|---|---|
| P1 | Spring | 39,941,799 | SCYLV-BRA | 348,348 | 1478 | 0.87 | 99 | 99 |
| SCMV | 1,722,495 | 4506 | 4.31 | 100 | 96 | |||
| P1 | Fall | 34,084,801 | SCYLV-BRA | 79,964 | 398 | 0.23 | 99 | 99 |
| SCMV | 837,377 | 2567 | 2.46 | 100 | 96 | |||
| P2 | Spring | 27,907,075 | SCYLV-CHN | 275,879 | 1682 | 0.99 | 100 | 99 |
| P2 | Fall | 25,794,227 | SCYLV-CHN | 41,048 | 271 | 0.16 | 99 | 99 |
| P3 | Spring | 26,379,182 | SCSMaV | 251,296 | 1169 | 0.95 | 100 | 98 |
| P3 | Fall | 42,730,577 | SCSMaV | 438,761 | 1261 | 1.03 | 100 | 98 |
| P4 | Spring | 24,572,155 | FDV | 217,508 | 1953 | 0.89 | 100 | 99 |
| P4 | Fall | 40,623,810 | FDV | 15,436 | 84 | 0.04 | 100 | 99 |
| P5 | Spring | 24,479,398 | SCSEV | 6414 | 97 | 0.03 | 100 | 98 |
| SCWSV | 1201 | 17 | 4.91 × 10−3 | 94 | 92 | |||
| P5 | Fall | 25,615,770 | SCSEV | 593 | 9 | 2.31 × 10−3 | 100 | 98 |
| SCWSV | 337 | 5 | 1.30 × 10−3 | 79 | 89 | |||
| P6 | Spring | 24,923,563 | SCYLV-CHN | 240,579 | 1642 | 0.97 | 100 | 99 |
| P6 | Fall | 25,630,762 | SCYLV-CHN | 69,202 | 459 | 0.27 | 99 | 99 |
| P7 | Spring | 27,901,105 | SCSMV | 1,497,825 | 5488 | 5.37 | 100 | 99 |
| P7 | Fall | 39,850,078 | SCSMV | 838,923 | 2152 | 2.11 | 100 | 99 |
| P8 | Spring | 32,773,246 | SCYLV-BRA | 211,597 | 1094 | 0.65 | 99 | 99 |
| P8 | Fall | 36,321,988 | SCYLV-BRA | 58,126 | 271 | 0.16 | 100 | 98 |
| P9 | Spring | 31,413,886 | SCYLV-BRA | 289,497 | 1562 | 0.92 | 100 | 99 |
| P9 | Fall | 39,661,498 | SCYLV-BRA | 21,790 | 93 | 0.05 | 99 | 99 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malapi-Wight, M.; Adhikari, B.; Zhou, J.; Hendrickson, L.; Maroon-Lango, C.J.; McFarland, C.; Foster, J.A.; Hurtado-Gonzales, O.P. HTS-Based Diagnostics of Sugarcane Viruses: Seasonal Variation and Its Implications for Accurate Detection. Viruses 2021, 13, 1627. https://doi.org/10.3390/v13081627
Malapi-Wight M, Adhikari B, Zhou J, Hendrickson L, Maroon-Lango CJ, McFarland C, Foster JA, Hurtado-Gonzales OP. HTS-Based Diagnostics of Sugarcane Viruses: Seasonal Variation and Its Implications for Accurate Detection. Viruses. 2021; 13(8):1627. https://doi.org/10.3390/v13081627
Chicago/Turabian StyleMalapi-Wight, Martha, Bishwo Adhikari, Jing Zhou, Leticia Hendrickson, Clarissa J. Maroon-Lango, Clint McFarland, Joseph A. Foster, and Oscar P. Hurtado-Gonzales. 2021. "HTS-Based Diagnostics of Sugarcane Viruses: Seasonal Variation and Its Implications for Accurate Detection" Viruses 13, no. 8: 1627. https://doi.org/10.3390/v13081627
APA StyleMalapi-Wight, M., Adhikari, B., Zhou, J., Hendrickson, L., Maroon-Lango, C. J., McFarland, C., Foster, J. A., & Hurtado-Gonzales, O. P. (2021). HTS-Based Diagnostics of Sugarcane Viruses: Seasonal Variation and Its Implications for Accurate Detection. Viruses, 13(8), 1627. https://doi.org/10.3390/v13081627