
New Insights into Human Cytomegalovirus pUL52 Structure

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification of Conserved Patterns and Structure Prediction
2.2. Cells and Bacterial Strains
2.3. HCMV Strains and Isolates
2.4. Amplification and Sequencing of the UL52 Gene from Reference Strains and Isolates
2.5. Heatmap Generation
2.6. pUL32 HSV-1 Polymorphism Study
2.7. Bacterial Artificial Chromosome Mutagenesis
2.8. Reconstitution of HCMV-BAC Viruses Harboring the Mutations
2.9. Plaque Assays and Growth Curve Analysis
2.10. Viral Immediate–Early and Late Proteins Expression
3. Results
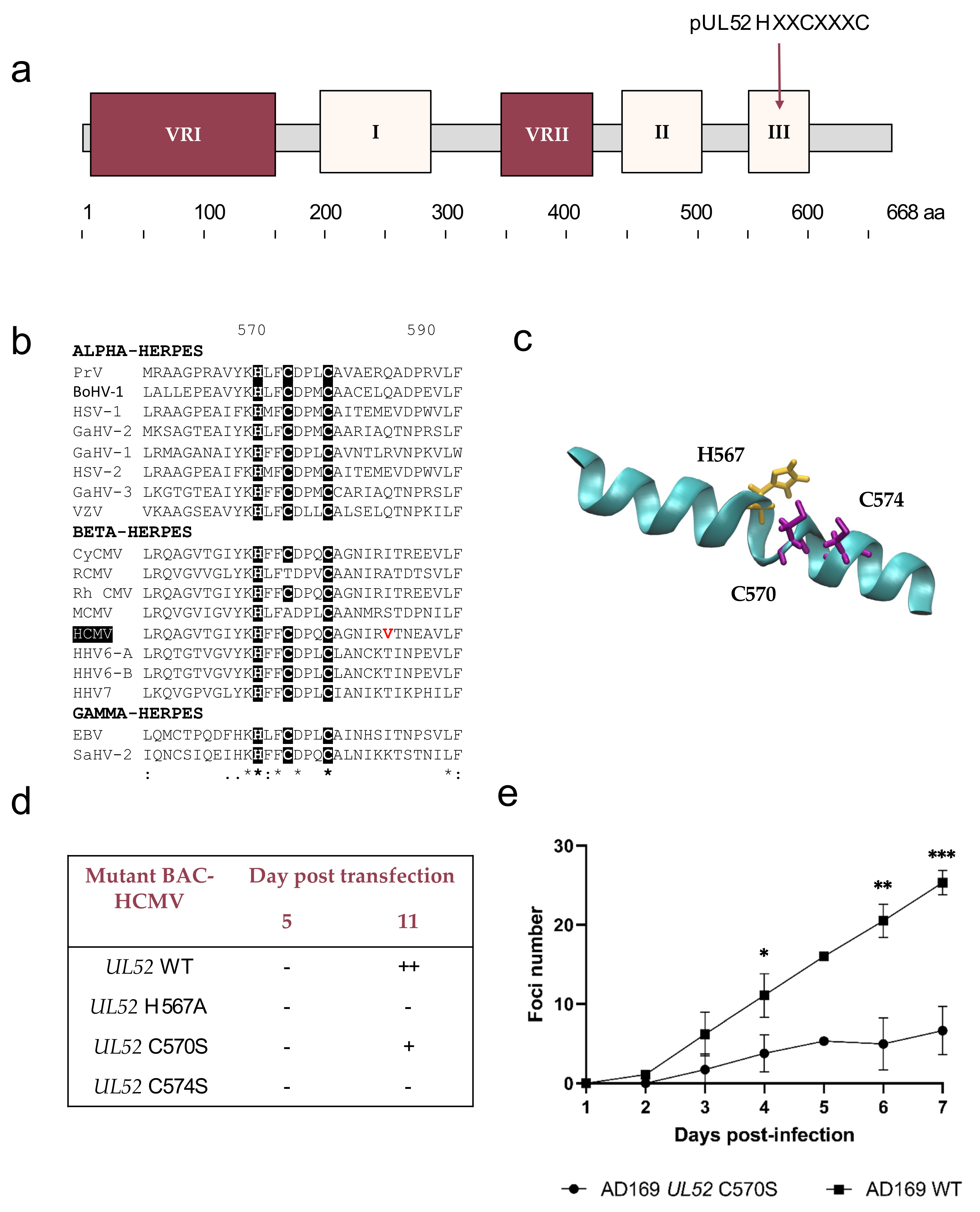
3.1. Identification of Conserved Regions
3.2. Identification of Sequence Polymorphisms
3.3. pUL32 Polymorphism Is Consistent with pUL52
3.4. Analysis of Conserved Region I Revealed Putative Metal-Binding Sites
3.5. Analysis of Conserved Region II
3.6. Analysis of Conserved Region III
3.7. pUL52 Mutations Do Not Impact the Viral Gene Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rubin, R.H. Prevention of Cytomegalovirus Infection in Organ Transplant Recipients. Transpl. Infect. Dis. 2000, 2, 99–100. [Google Scholar] [CrossRef] [PubMed]
- Leruez-Ville, M.; Ville, Y. Fetal Cytomegalovirus Infection. Best Pract. Res. Clin. Obs. Gynaecol. 2017, 38, 97–107. [Google Scholar] [CrossRef]
- Lurain, N.S.; Chou, S. Antiviral Drug Resistance of Human Cytomegalovirus. Clin. Microbiol. Rev. 2010, 23, 689–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Chaer, F.; Shah, D.P.; Chemaly, R.F. How I Treat Resistant Cytomegalovirus Infection in Hematopoietic Cell Transplantation Recipients. Blood 2016, 128, 2624–2636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andouard, D.; Mazeron, M.-C.; Ligat, G.; Couvreux, A.; Pouteil-Noble, C.; Cahen, R.; Yasdanpanah, Y.; Deering, M.; Viget, N.; Alain, S.; et al. Contrasting Effect of New HCMV PUL54 Mutations on Antiviral Drug Susceptibility: Benefits and Limits of 3D Analysis. Antivir. Res. 2016, 129, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Marty, F.M.; Ljungman, P.; Chemaly, R.F.; Maertens, J.; Dadwal, S.S.; Duarte, R.F.; Haider, S.; Ullmann, A.J.; Katayama, Y.; Brown, J.; et al. Letermovir Prophylaxis for Cytomegalovirus in Hematopoietic-Cell Transplantation. N. Engl. J. Med. 2017, 377, 2433–2444. [Google Scholar] [CrossRef]
- Chou, S.; Satterwhite, L.E.; Ercolani, R.J. New Locus of Drug Resistance in the Human Cytomegalovirus UL56 Gene Revealed by In Vitro Exposure to Letermovir and Ganciclovir. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, S. A Third Component of the Human Cytomegalovirus Terminase Complex Is Involved in Letermovir Resistance. Antivir. Res. 2017, 148, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Ligat, G.; Cazal, R.; Hantz, S.; Alain, S. The Human Cytomegalovirus Terminase Complex as an Antiviral Target: A Close-up View. FEMS Microbiol. Rev. 2018, 42, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Muller, C.; Alain, S.; Baumert, T.F.; Ligat, G.; Hantz, S. Structures and Divergent Mechanisms in Capsid Maturation and Stabilization Following Genome Packaging of Human Cytomegalovirus and Herpesviruses. Life 2021, 11, 150. [Google Scholar] [CrossRef]
- Borst, E.M.; Kleine-Albers, J.; Gabaev, I.; Babić, M.; Wagner, K.; Binz, A.; Degenhardt, I.; Kalesse, M.; Jonjić, S.; Bauerfeind, R.; et al. The Human Cytomegalovirus UL51 Protein Is Essential for Viral Genome Cleavage-Packaging and Interacts with the Terminase Subunits PUL56 and PUL89. J. Virol. 2013, 87, 1720–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borst, E.M.; Wagner, K.; Binz, A.; Sodeik, B.; Messerle, M. The Essential Human Cytomegalovirus Gene UL52 Is Required for Cleavage-Packaging of the Viral Genome. J. Virol. 2008, 82, 2065–2078. [Google Scholar] [CrossRef] [Green Version]
- Borst, E.M.; Bauerfeind, R.; Binz, A.; Stephan, T.M.; Neuber, S.; Wagner, K.; Steinbrück, L.; Sodeik, B.; Lenac Roviš, T.; Jonjić, S.; et al. The Essential Human Cytomegalovirus Proteins PUL77 and PUL93 Are Structural Components Necessary for Viral Genome Encapsidation. J. Virol. 2016, 90, 5860–5875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeRussy, B.M.; Tandon, R. Human Cytomegalovirus PUL93 Is Required for Viral Genome Cleavage and Packaging. J. Virol. 2015, 89, 12221–12225. [Google Scholar] [CrossRef] [Green Version]
- Köppen-Rung, P.; Dittmer, A.; Bogner, E. Intracellular Distribution of Capsid-Associated PUL77 of Human Cytomegalovirus and Interactions with Packaging Proteins and PUL93. J. Virol. 2016, 90, 5876–5885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albright, B.S.; Kosinski, A.; Szczepaniak, R.; Cook, E.A.; Stow, N.D.; Conway, J.F.; Weller, S.K. The Putative Herpes Simplex Virus 1 Chaperone Protein UL32 Modulates Disulfide Bond Formation during Infection. J. Virol. 2015, 89, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.E.; Poon, A.P.; Roizman, B. Properties of the Protein Encoded by the UL32 Open Reading Frame of Herpes Simplex Virus 1. J. Virol. 1996, 70, 3938–3946. [Google Scholar] [CrossRef] [Green Version]
- Lamberti, C.; Weller, S.K. The Herpes Simplex Virus Type 1 Cleavage/Packaging Protein, UL32, Is Involved in Efficient Localization of Capsids to Replication Compartments. J. Virol. 1998, 72, 2463–2473. [Google Scholar] [CrossRef] [Green Version]
- Giesen, K.; Radsak, K.; Bogner, E. The Potential Terminase Subunit of Human Cytomegalovirus, PUL56, Is Translocated into the Nucleus by Its Own Nuclear Localization Signal and Interacts with Importin α. J. Gen. Virol. 2000, 81, 2231–2244. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-Quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- McWilliam, H.; Li, W.; Uludag, M.; Squizzato, S.; Park, Y.M.; Buso, N.; Cowley, A.P.; Lopez, R. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 2013, 41, W597–W600. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Cowley, A.; Uludag, M.; Gur, T.; McWilliam, H.; Squizzato, S.; Park, Y.M.; Buso, N.; Lopez, R. The EMBL-EBI Bioinformatics Web and Programmatic Tools Framework. Nucleic Acids Res. 2015, 43, W580–W584. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A Unified Platform for Automated Protein Structure and Function Prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein Structure and Function Prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhang, Y. I-TASSER Server: New Development for Protein Structure and Function Predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borst, E.-M.; Hahn, G.; Koszinowski, U.H.; Messerle, M. Cloning of the Human Cytomegalovirus (HCMV) Genome as an Infectious Bacterial Artificial Chromosome in Escherichia Coli: A New Approach for Construction of HCMV Mutants. J. Virol. 1999, 73, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chee, M.S.; Bankier, A.T.; Beck, S.; Bohni, R.; Brown, C.M.; Cerny, R.; Horsnell, T.; Hutchison, C.A.; Kouzarides, T.; Martignetti, J.A. Analysis of the Protein-Coding Content of the Sequence of Human Cytomegalovirus Strain AD169. Curr. Top. Microbiol. Immunol. 1990, 154, 125–169. [Google Scholar] [CrossRef] [PubMed]
- Ligat, G.; Jacquet, C.; Chou, S.; Couvreux, A.; Alain, S.; Hantz, S. Identification of a Short Sequence in the HCMV Terminase PUL56 Essential for Interaction with PUL89 Subunit. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Hirt, B. Selective Extraction of Polyoma DNA from Infected Mouse Cell Cultures. J. Mol. Biol. 1967, 26, 365–369. [Google Scholar] [CrossRef]
- Ligat, G.; Couvreux, A.; Cazal, R.; Alain, S.; Hantz, S. Highlighting of a LAGLIDADG and a Zing Finger Motifs Located in the PUL56 Sequence Crucial for HCMV Replication. Viruses 2019, 11, 1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Champier, G.; Couvreux, A.; Hantz, S.; Rametti, A.; Mazeron, M.-C.; Bouaziz, S.; Denis, F.; Alain, S. Original Article Putative Functional Domains of Human Cytomegalovirus PUL56 Involved in Dimerization and Benzimidazole D-Ribonucleoside Activity. Antivir. Ther. 2008, 13, 643–654. [Google Scholar] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muller, C.; Alain, S.; Gourin, C.; Baumert, T.F.; Ligat, G.; Hantz, S. New Insights into Human Cytomegalovirus pUL52 Structure. Viruses 2021, 13, 1638. https://doi.org/10.3390/v13081638
Muller C, Alain S, Gourin C, Baumert TF, Ligat G, Hantz S. New Insights into Human Cytomegalovirus pUL52 Structure. Viruses. 2021; 13(8):1638. https://doi.org/10.3390/v13081638
Chicago/Turabian StyleMuller, Clotilde, Sophie Alain, Claire Gourin, Thomas F. Baumert, Gaëtan Ligat, and Sébastien Hantz. 2021. "New Insights into Human Cytomegalovirus pUL52 Structure" Viruses 13, no. 8: 1638. https://doi.org/10.3390/v13081638
APA StyleMuller, C., Alain, S., Gourin, C., Baumert, T. F., Ligat, G., & Hantz, S. (2021). New Insights into Human Cytomegalovirus pUL52 Structure. Viruses, 13(8), 1638. https://doi.org/10.3390/v13081638