
APOBEC Mutagenesis Is Concordant between Tumor and Viral Genomes in HPV-Positive Head and Neck Squamous Cell Carcinoma

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Sequencing
2.3. Informatics
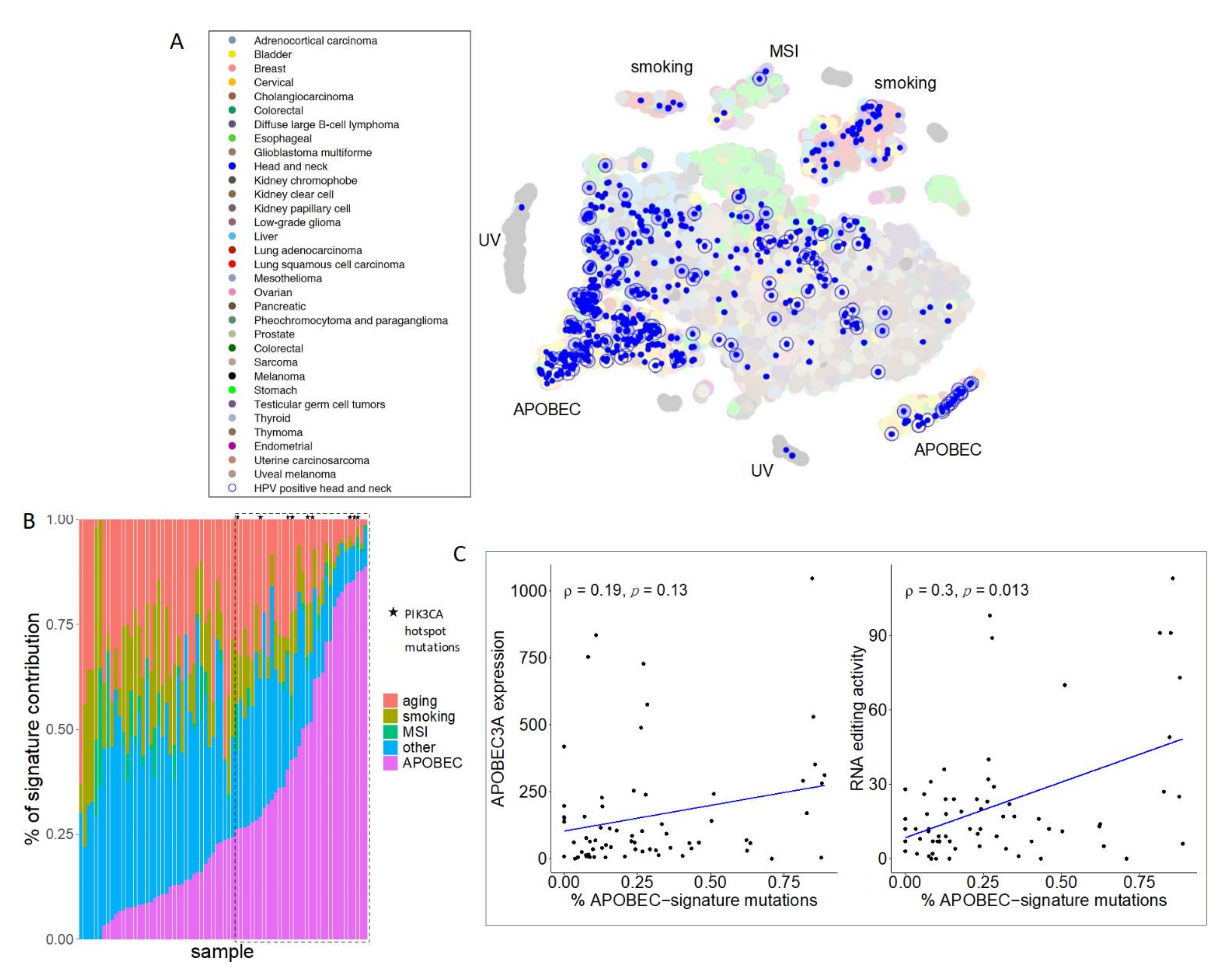
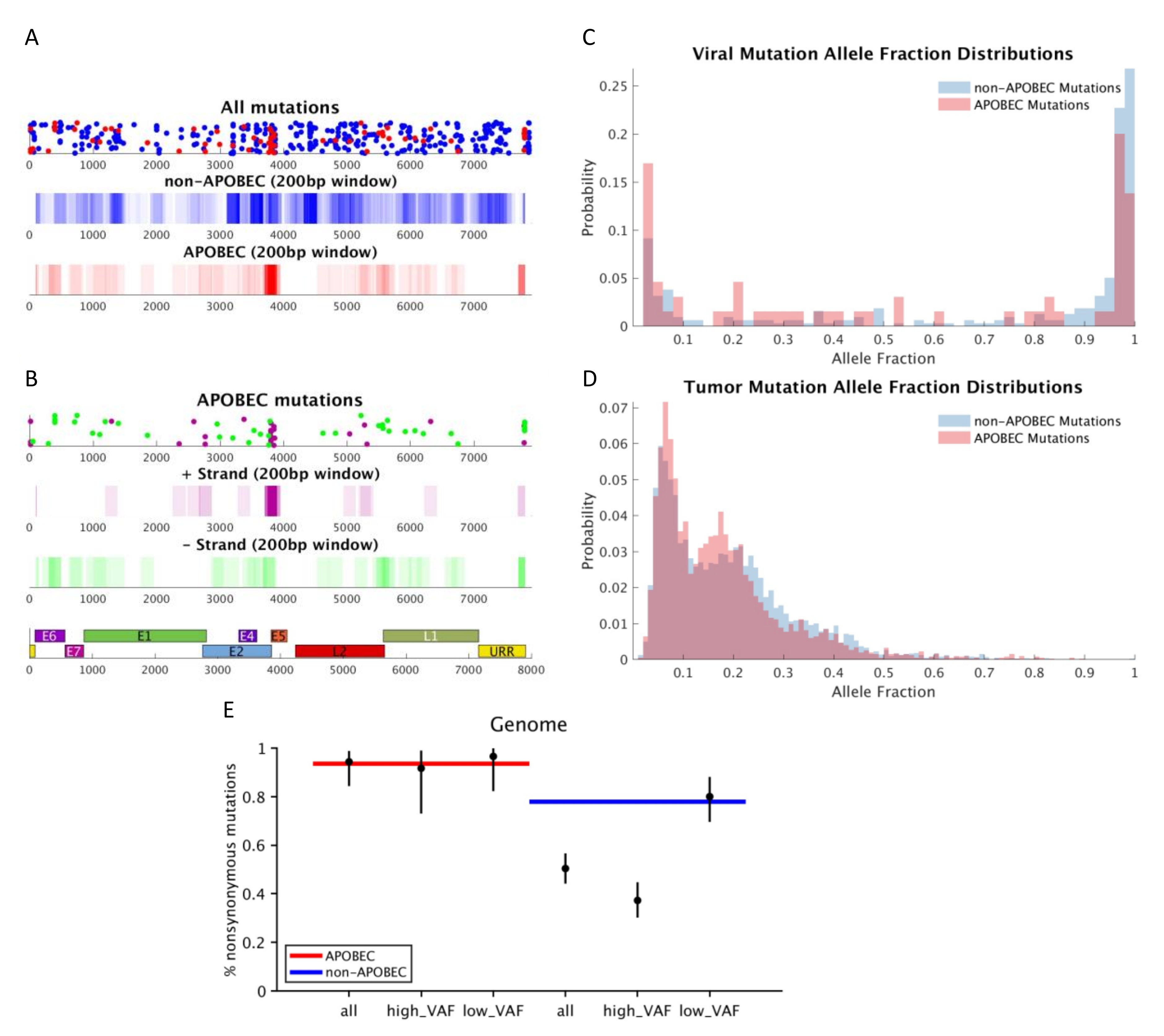
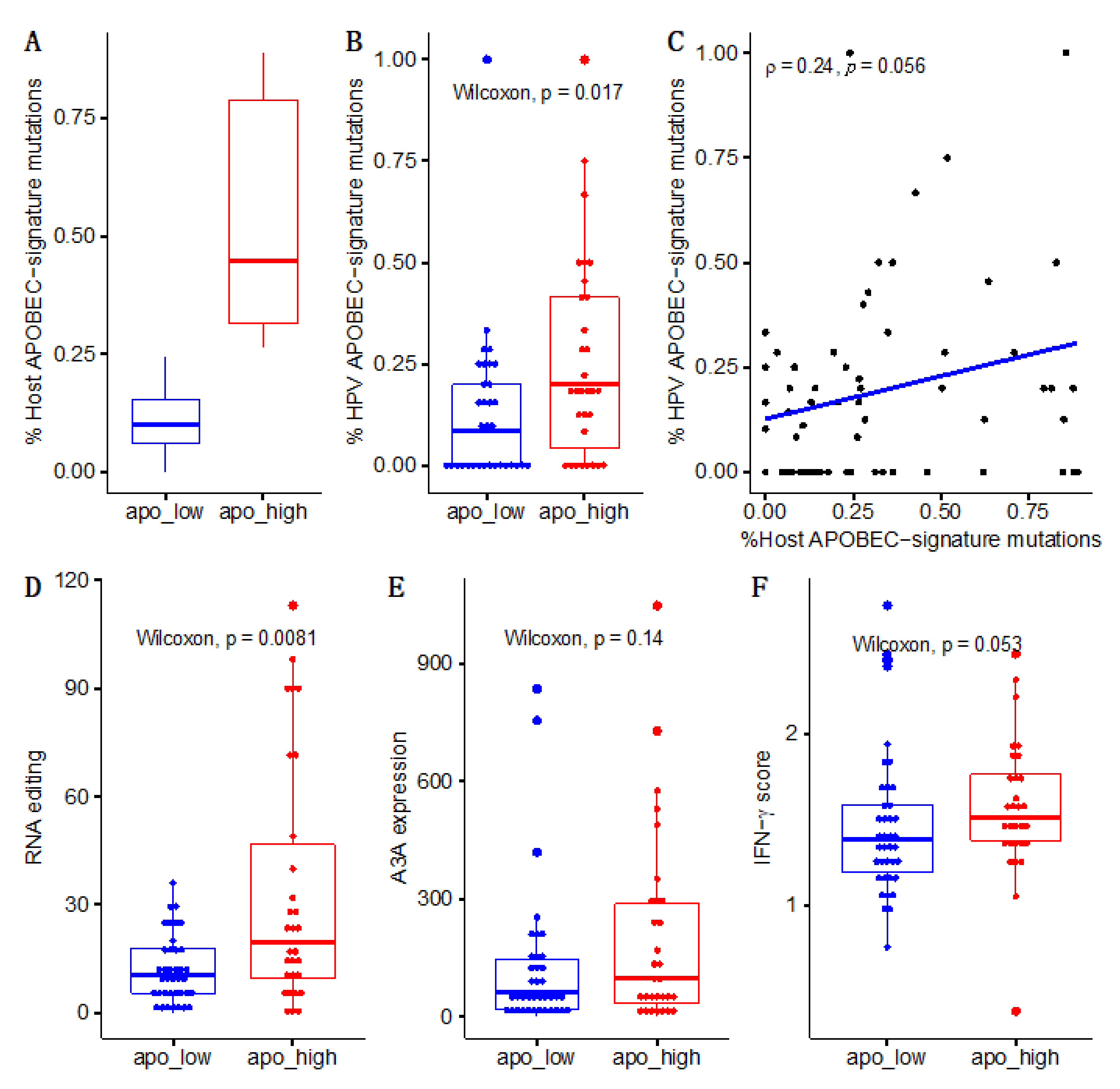
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.V.; Carter, S.L.; Saksena, G.; et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar] [CrossRef]
- Henderson, S.; Chakravarthy, A.; Su, X.; Boshoff, C.; Fenton, T.R. APOBEC-mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus-driven tumor development. Cell Rep. 2014, 7, 1833–1841. [Google Scholar] [CrossRef] [Green Version]
- Faden, D.L.; Thomas, S.; Cantalupo, P.G.; Agrawal, N.; Myers, J.; DeRisi, J. Multi-modality analysis supports APOBEC as a major source of mutations in head and neck squamous cell carcinoma. Oral Oncol. 2017, 74, 8–14. [Google Scholar] [CrossRef]
- Faden, D.L.; Langenbucher, A.; Kuhs, K.; Lewis, J.S.; Mirabello, L.; Yeager, M.; Boland, J.F.; Bass, S.; Steinberg, M.; Cullen, M.; et al. HPV+ Oropharyngeal Squamous Cell Carcinomas from patients with two tumors display synchrony of viral genomes yet discordant mutational profiles and signatures. Carcinogenesis 2020, 42, 14–20. [Google Scholar] [CrossRef]
- Faden, D.L.; Ding, F.; Lin, Y.; Zhai, S.; Kuo, F.; Chan, T.A.; Morris, L.G.; Ferris, R.L. APOBEC mutagenesis is tightly linked to the immune landscape and immunotherapy biomarkers in head and neck squamous cell carcinoma. Oral Oncol. 2019, 96, 140–147. [Google Scholar] [CrossRef]
- Smith, N.J.; Fenton, T.R. The APOBEC3 genes and their role in cancer: Insights from human papillomavirus. J. Mol. Endocrinol. 2019, 62, R269–R287. [Google Scholar] [CrossRef] [Green Version]
- Mori, S.; Takeuchi, T.; Ishii, Y.; Kukimoto, I. Identification of APOBEC3B promoter elements responsible for activation by human papillomavirus type 16 E6. Biochem. Biophys. Res. Commun. 2015, 460, 555–560. [Google Scholar] [CrossRef]
- Vieira, V.C.; Leonard, B.; White, E.A.; Starrett, G.J.; Temiz, N.A.; Lorenz, L.D.; Lee, D.; Soares, M.A.; Lambert, P.F.; Howley, P.M.; et al. Human papillomavirus E6 triggers upregulation of the antiviral and cancer genomic DNA deaminase APOBEC3B. MBio 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wakae, K.; Kitamura, K.; Aoyama, S.; Liu, G.; Koura, M.; Monjurul, A.M.; Kukimoto, I.; Muramatsu, M. APOBEC3 deaminases induce hypermutation in human papillomavirus 16 DNA upon beta interferon stimulation. J. Virol. 2014, 88, 1308–1317. [Google Scholar] [CrossRef] [Green Version]
- Warren, C.J.; Xu, T.; Guo, K.; Griffin, L.M.; Westrich, J.A.; Lee, D.; Lambert, P.F.; Santiago, M.L.; Pyeon, D. APOBEC3A functions as a restriction factor of human papillomavirus. J. Virol. 2015, 89, 688–702. [Google Scholar] [CrossRef] [Green Version]
- Westrich, J.A.; Warren, C.J.; Klausner, M.J.; Guo, K.; Liu, C.W.; Santiago, M.L.; Pyeon, D. Human Papillomavirus 16 E7 Stabilizes APOBEC3A Protein by Inhibiting Cullin 2-Dependent Protein Degradation. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Vartanian, J.P.; Guetard, D.; Henry, M.; Wain-Hobson, S. Evidence for editing of human papillomavirus DNA by APOBEC3 in benign and precancerous lesions. Science 2008, 320, 230–233. [Google Scholar] [CrossRef] [Green Version]
- Wakae, K.; Aoyama, S.; Wang, Z.; Kitamura, K.; Liu, G.; Monjurul, A.M.; Koura, M.; Imayasu, M.; Sakamoto, N.; Nakamura, M.; et al. Detection of hypermutated human papillomavirus type 16 genome by Next-Generation Sequencing. Virology 2015, 485, 460–466. [Google Scholar] [CrossRef] [Green Version]
- Kukimoto, I.; Mori, S.; Aoyama, S.; Wakae, K.; Muramatsu, M.; Kondo, K. Hypermutation in the E2 gene of human papillomavirus type 16 in cervical intraepithelial neoplasia. J. Med. Virol. 2015, 87, 1754–1760. [Google Scholar] [CrossRef]
- Hirose, Y.; Onuki, M.; Tenjimbayashi, Y.; Mori, S.; Ishii, Y.; Takeuchi, T.; Tasaka, N.; Satoh, T.; Morisada, T.; Iwata, T.; et al. Within-Host Variations of Human Papillomavirus Reveal APOBEC Signature Mutagenesis in the Viral Genome. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Gillison, M.L.; Akagi, K.; Xiao, W.; Jiang, B.; Pickard, R.K.L.; Li, J.; Swanson, B.J.; Agrawal, A.D.; Zucker, M.; Stache-Crain, B.; et al. Human papillomavirus and the landscape of secondary genetic alterations in oral cancers. Genome Res. 2019, 29, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Seplyarskiy, V.B.; Soldatov, R.A.; Popadin, K.Y.; Antonarakis, S.E.; Bazykin, G.A.; Nikolaev, S.I. APOBEC-induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication. Genome Res. 2016, 26, 174–182. [Google Scholar] [CrossRef] [Green Version]
- Swanton, C.; McGranahan, N.; Starrett, G.J.; Harris, R.S. APOBEC Enzymes: Mutagenic Fuel for Cancer Evolution and Heterogeneity. Cancer Discov. 2015, 5, 704–712. [Google Scholar] [CrossRef] [Green Version]
- Burns, M.B.; Temiz, N.A.; Harris, R.S. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 2013, 45, 977–983. [Google Scholar] [CrossRef] [Green Version]
- Hildesheim, A.; Schiffman, M.; Bromley, C.; Wacholder, S.; Herrero, R.; Rodriguez, A.C.; Bratti, M.C.; Sherman, M.E.; Scarpidis, U.; Lin, Q.-Q.; et al. Human Papillomavirus Type 16 Variants and Risk of Cervical Cancer. J. Natl. Cancer Inst. 2001, 93, 315–318. [Google Scholar] [CrossRef] [Green Version]
- Pientong, C.; Wongwarissara, P.; Ekalaksananan, T.; Swangphon, P.; Kleebkaow, P.; Kongyingyoes, B.; Siriaunkgul, S.; Tungsinmunkong, K.; Suthipintawong, C. Association of human papillomavirus type 16 long control region mutation and cervical cancer. Virol. J. 2013, 10, 30. [Google Scholar]
- Xi, L.F.; Koutsky, L.A.; Hildesheim, A.; Galloway, D.A.; Wheeler, C.M.; Winer, R.L.; Ho, J.; Kiviat, N.B. Risk for High-Grade Cervical Intraepithelial Neoplasia Associated with Variants of Human Papillomavirus Types 16 and 18. Cancer Epidemiol. Biomark. Prev. 2007, 16, 4–10. [Google Scholar] [CrossRef] [Green Version]
- Schiffman, M.; Rodriguez, A.C.; Chen, Z.; Wacholder, S.; Herrero, R.; Hildesheim, A.; Desalle, R.; Befano, B.; Yu, K.; Safaeian, M.; et al. A Population-Based Prospective Study of Carcinogenic Human Papillomavirus Variant Lineages, Viral Persistence, and Cervical Neoplasia. Cancer Res. 2010, 70, 3159–3169. [Google Scholar] [CrossRef] [Green Version]
- Cornet, I.; Gheit, T.; Iannacone, M.R.; Vignat, J.; Sylla, B.S.; Del Mistro, A.; Franceschi, S.; Tommasino, M.; Clifford, G.M. HPV16 genetic variation and the development of cervical cancer worldwide. Br. J. Cancer 2013, 108, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Gheit, T.; Cornet, I.; Clifford, G.M.; Iftner, T.; Munk, C.; Tommasino, M.; Kjaer, S.K. Risks for Persistence and Progression by Human Papillomavirus Type 16 Variant Lineages Among a Population-Based Sample of Danish Women. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1315–1321. [Google Scholar] [CrossRef] [Green Version]
- Zehbe, I.; Voglino, G.; Delius, H.; Wilander, E.; Tommasino, M. Risk of cervical cancer and geographical variations of human papillomavirus 16 E6 polymorphisms. Lancet 1998, 352, 1441–1442. [Google Scholar] [CrossRef]
- Zuna, R.E.; Moore, W.E.; Shanesmith, R.P.; Dunn, S.T.; Wang, S.S.; Schiffman, M.; Blakey, G.L.; Teel, T. Association of HPV16 E6 variants with diagnostic severity in cervical cytology samples of 354 women in a US population. Int. J. Cancer 2009, 125, 2609–2613. [Google Scholar] [CrossRef] [Green Version]
- Sichero, L.; Ferreira, S.; Trottier, H.; Duarte-Franco, E.; Ferenczy, A.; Franco, E.L.; Villa, L.L. High grade cervical lesions are caused preferentially by non-European variants of HPVs 16 and 18. Int. J. Cancer 2007, 120, 1763–1768. [Google Scholar] [CrossRef]
- Berumen, J.; Ordoñez, R.M.; Lazcano, E.; Salmeron, J.; Galvan, S.C.; Estrada, R.A.; Yunes, E.; Garcia-Carranca, A.; Gonzalez-Lira, G.; Madrigal-de la Campa, A. Asian-American Variants of Human Papillomavirus 16 and Risk for Cervical Cancer: A Case–Control Study. J. Natl. Cancer Inst. 2001, 93, 1325–1330. [Google Scholar] [CrossRef]
- Freitas, L.B.; Chen, Z.; Muqui, E.F.; Boldrini, N.A.T.; Miranda, A.E.; Spano, L.C.; Burk, R.D. Human Papillomavirus 16 Non-European Variants Are Preferentially Associated with High-Grade Cervical Lesions. PLoS ONE 2014, 9, e100746. [Google Scholar] [CrossRef] [Green Version]
- Mirabello, L.; Yeager, M.; Cullen, M.; Boland, J.F.; Chen, Z.; Wentzensen, N.; Zhang, X.; Yu, K.; Yang, Q.; Mitchell, J.; et al. HPV16 Sublineage Associations With Histology-Specific Cancer Risk Using HPV Whole-Genome Sequences in 3200 Women. J. Natl. Cancer Inst. 2016, 108, djw100. [Google Scholar]
- Mirabello, L.; Clarke, M.A.; Nelson, C.W.; Dean, M.; Wentzensen, N.; Yeager, M.; Cullen, M.; Boland, J.F.; Workshop, N.H.; Schiffman, M.; et al. The Intersection of HPV Epidemiology, Genomics and Mechanistic Studies of HPV-Mediated Carcinogenesis. Viruses 2018, 10, 80. [Google Scholar] [CrossRef] [Green Version]
- Mirabello, L.; Yeager, M.; Yu, K.; Clifford, G.M.; Xiao, Y.; Zhu, B.; Cullen, M.; Boland, J.F.; Wentzensen, N.; Nelson, C.W.; et al. HPV16 E7 Genetic Conservation Is Critical to Carcinogenesis. Cell 2017, 170, 1164–1174.e1166. [Google Scholar] [CrossRef] [Green Version]
- Pinheiro, M.; Gage, J.C.; Clifford, G.M.; Demarco, M.; Cheung, L.C.; Chen, Z.; Yeager, M.; Cullen, M.; Boland, J.F.; Chen, X.; et al. Association of HPV35 with cervical carcinogenesis among women of African ancestry: Evidence of viral-host interaction with implications for disease intervention. Int. J. Cancer 2020, 147, 2677–2686. [Google Scholar] [CrossRef]
- Zhu, B.; Xiao, Y.; Yeager, M.; Clifford, G.; Wentzensen, N.; Cullen, M.; Boland, J.F.; Bass, S.; Steinberg, M.K.; Raine-Bennett, T.; et al. Mutations in the HPV16 genome induced by APOBEC3 are associated with viral clearance. Nat. Commun. 2020, 11, 886. [Google Scholar] [CrossRef] [Green Version]
- Cullen, M.; Boland, J.F.; Schiffman, M.; Zhang, X.; Wentzensen, N.; Yang, Q.; Chen, Z.; Yu, K.; Mitchell, J.; Roberson, D.; et al. Deep sequencing of HPV16 genomes: A new high-throughput tool for exploring the carcinogenicity and natural history of HPV16 infection. Papillomavirus Res. 2015, 1, 3–11. [Google Scholar] [CrossRef] [Green Version]
- TCGA. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef]
- Fang, W.L.; Huang, K.H.; Lan, Y.T.; Lin, C.H.; Chang, S.C.; Chen, M.H.; Chao, Y.; Lin, W.C.; Lo, S.S.; Li, A.F.; et al. Mutations in PI3K/AKT pathway genes and amplifications of PIK3CA are associated with patterns of recurrence in gastric cancers. Oncotarget 2016, 7, 6201–6220. [Google Scholar] [CrossRef] [Green Version]
- Van Doorslaer, K.; Li, Z.; Xirasagar, S.; Maes, P.; Kaminsky, D.; Liou, D.; Sun, Q.; Kaur, R.; Huyen, Y.; McBride, A.A. The Papillomavirus Episteme: A major update to the papillomavirus sequence database. Nucleic Acids Res. 2016, 45, D499–D506. [Google Scholar] [CrossRef]
- Jalili, P.; Bowen, D.; Langenbucher, A.; Park, S.; Aguirre, K.; Corcoran, R.B.; Fleischman, A.G.; Lawrence, M.S.; Zou, L.; Buisson, R. Quantification of ongoing APOBEC3A activity in tumor cells by monitoring RNA editing at hotspots. Nat. Commun. 2020, 11, 2971. [Google Scholar] [CrossRef]
- Brown, C.; Kowalczyk, A.M.; Taylor, E.R.; Morgan, I.M.; Gaston, K. P53 represses human papillomavirus type 16 DNA replication via the viral E2 protein. Virol. J. 2008, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Haradhvala, N.J.; Polak, P.; Stojanov, P.; Covington, K.R.; Shinbrot, E.; Hess, J.M.; Rheinbay, E.; Kim, J.; Maruvka, Y.E.; Braunstein, L.Z.; et al. Mutational Strand Asymmetries in Cancer Genomes Reveal Mechanisms of DNA Damage and Repair. Cell 2016, 164, 538–549. [Google Scholar] [CrossRef] [Green Version]
- Warren, C.J.; Van Doorslaer, K.; Pandey, A.; Espinosa, J.M.; Pyeon, D. Role of the host restriction factor APOBEC3 on papillomavirus evolution. Virus Evol. 2015, 1. [Google Scholar] [CrossRef]
- McBride, A.A. The papillomavirus E2 proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faden, D.L.; Kuhs, K.A.L.; Lin, M.; Langenbucher, A.; Pinheiro, M.; Yeager, M.; Cullen, M.; Boland, J.F.; Steinberg, M.; Bass, S.; et al. APOBEC Mutagenesis Is Concordant between Tumor and Viral Genomes in HPV-Positive Head and Neck Squamous Cell Carcinoma. Viruses 2021, 13, 1666. https://doi.org/10.3390/v13081666
Faden DL, Kuhs KAL, Lin M, Langenbucher A, Pinheiro M, Yeager M, Cullen M, Boland JF, Steinberg M, Bass S, et al. APOBEC Mutagenesis Is Concordant between Tumor and Viral Genomes in HPV-Positive Head and Neck Squamous Cell Carcinoma. Viruses. 2021; 13(8):1666. https://doi.org/10.3390/v13081666
Chicago/Turabian StyleFaden, Daniel L., Krystle A. Lang Kuhs, Maoxuan Lin, Adam Langenbucher, Maisa Pinheiro, Meredith Yeager, Michael Cullen, Joseph F. Boland, Mia Steinberg, Sara Bass, and et al. 2021. "APOBEC Mutagenesis Is Concordant between Tumor and Viral Genomes in HPV-Positive Head and Neck Squamous Cell Carcinoma" Viruses 13, no. 8: 1666. https://doi.org/10.3390/v13081666
APA StyleFaden, D. L., Kuhs, K. A. L., Lin, M., Langenbucher, A., Pinheiro, M., Yeager, M., Cullen, M., Boland, J. F., Steinberg, M., Bass, S., Lewis, J. S., Lawrence, M. S., Ferris, R. L., & Mirabello, L. (2021). APOBEC Mutagenesis Is Concordant between Tumor and Viral Genomes in HPV-Positive Head and Neck Squamous Cell Carcinoma. Viruses, 13(8), 1666. https://doi.org/10.3390/v13081666