
Variability in Codon Usage in Coronaviruses Is Mainly Driven by Mutational Bias and Selective Constraints on CpG Dinucleotide

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Variation in Codon Usage Bias among Orthocoronavirinae Is Not Dependent on the Host
2.2. The Mutational Spectrum in Orthocoronavirinae Is AU-Biased
2.3. Recent Human Coronaviruses Display Greater Mutational Disequilibrium Than Endemic Human Coronaviruses
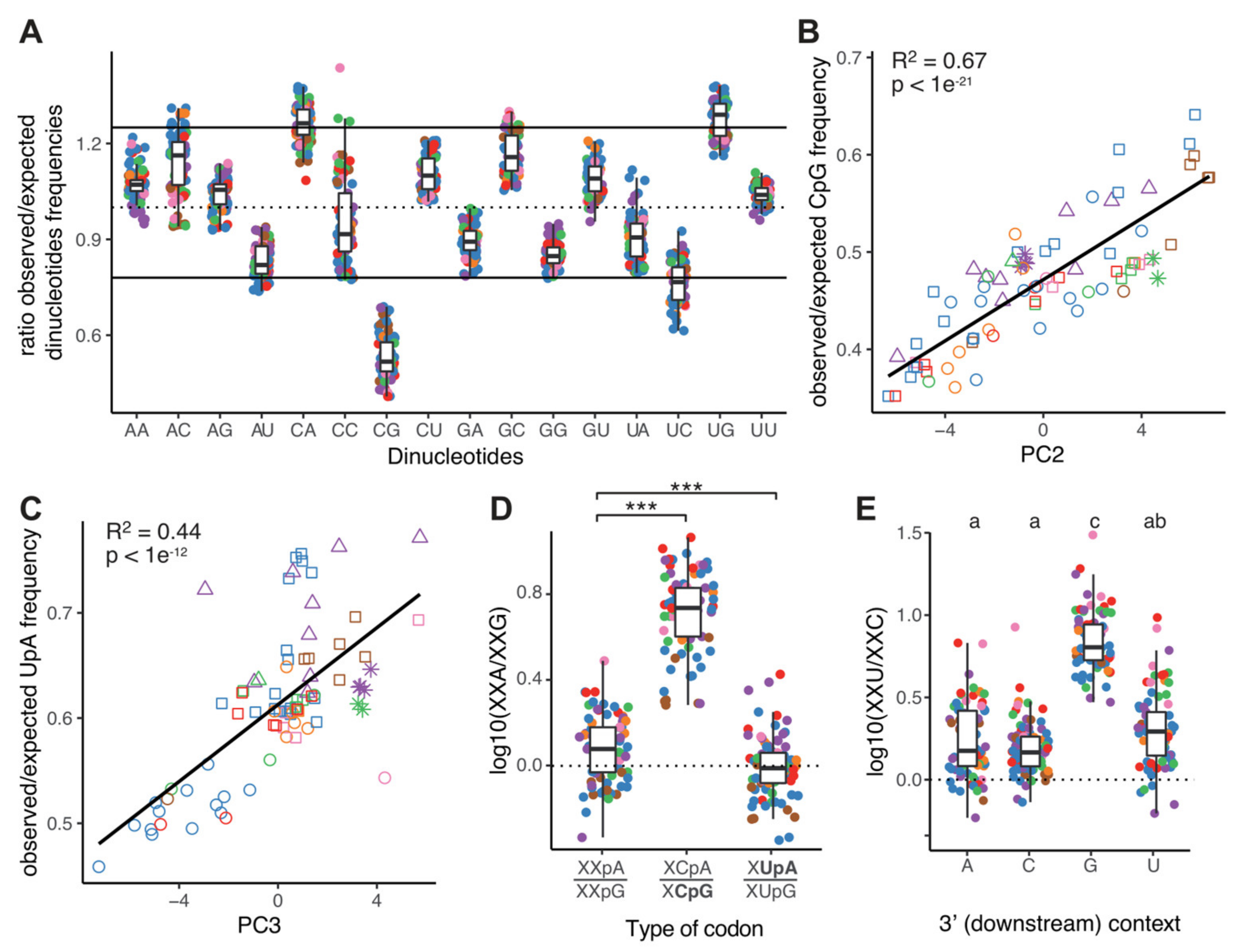
2.4. CpG Dinucleotides Are Selected against in Orthocoronavirinae Genomes
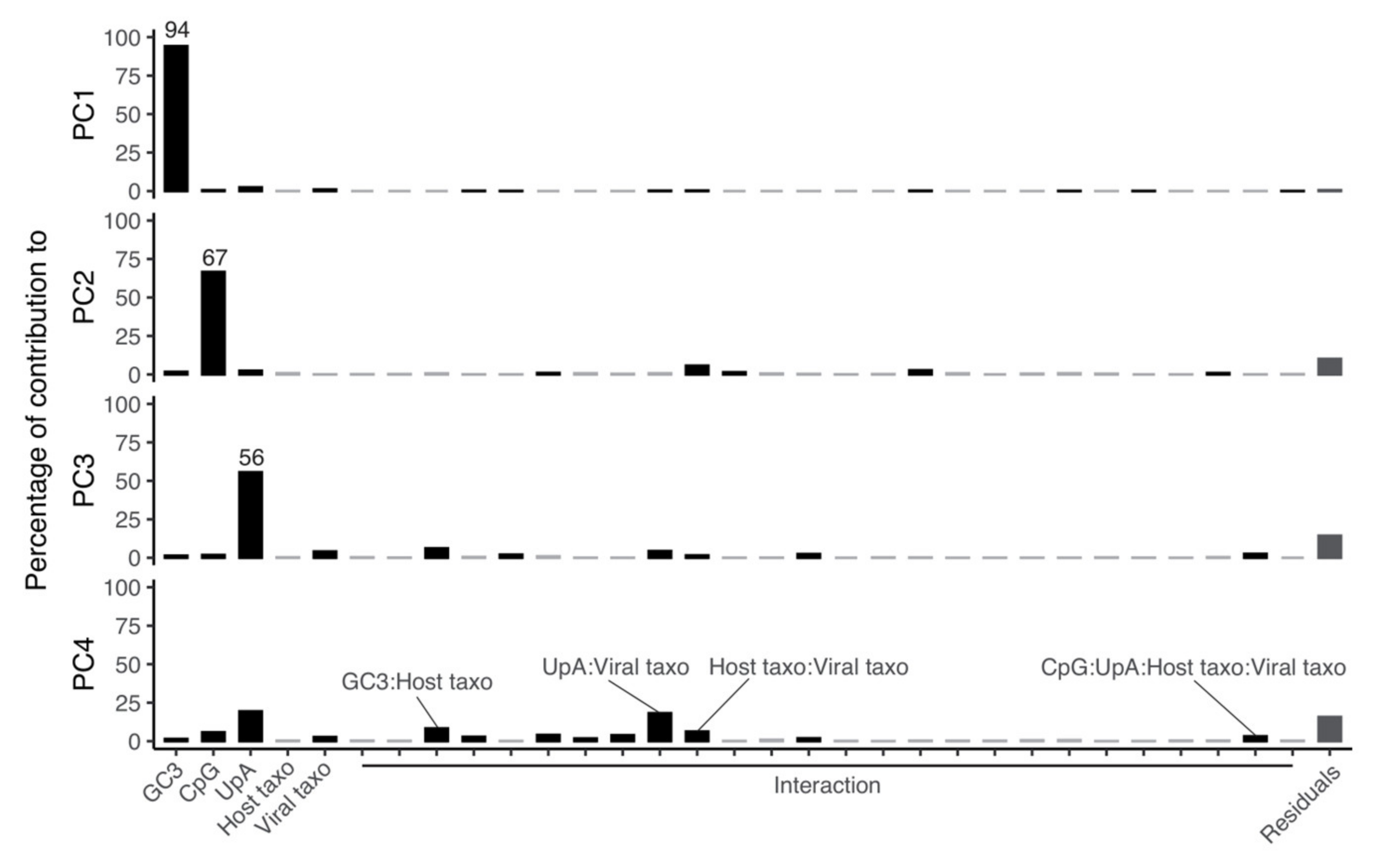
2.5. Mutational Bias and CpG/UpA Depletion Explain Most of the Variation in Synonymous Codon Usage of Coronaviruses
3. Discussion
3.1. Mutational Bias and CpG Avoidance Shape Codon Usage Bias in CoVs
3.2. Lack of Evidence for Translational Selection Acting on CoVs
3.3. Composition of CoVs Genomes Tends to Reach Their Mutational Equilibria
3.4. Conclusion: CUB Is a Poor Proxy to Predict Zoonotic Infection in CoVs
4. Material and Methods
4.1. Data Collection and Processing
4.2. Nucleotide Composition Analysis
4.3. SNP Calling
4.4. Assessment Mutation Profiles
4.5. Site Frequency Spectrum Assessment Mutational Equilibrium
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Boni, M.F.; Lemey, P.; Jiang, X.; Lam, T.T.-Y.; Perry, B.W.; Castoe, T.A.; Rambaut, A.; Robertson, D.L. Evolutionary Origins of the SARS-CoV-2 Sarbecovirus Lineage Responsible for the COVID-19 Pandemic. Nat. Microbiol. 2020, 5, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.L.; Baric, R.S. Recombination, Reservoirs, and the Modular Spike: Mechanisms of Coronavirus Cross-Species Transmission. J. Virol. 2010, 84, 3134–3146. [Google Scholar] [CrossRef]
- Forni, D.; Cagliani, R.; Clerici, M.; Sironi, M. Molecular Evolution of Human Coronavirus Genomes. Trends Microbiol. 2017, 25, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Anthony, S.J.; Johnson, C.K.; Greig, D.J.; Kramer, S.; Che, X.; Wells, H.; Hicks, A.L.; Joly, D.O.; Wolfe, N.D.; Daszak, P.; et al. Global Patterns in Coronavirus Diversity. Virus Evol. 2017, 3, vex012. [Google Scholar] [CrossRef]
- Leopardi, S.; Holmes, E.C.; Gastaldelli, M.; Tassoni, L.; Priori, P.; Scaravelli, D.; Zamperin, G.; De Benedictis, P. Interplay between Co-Divergence and Cross-Species Transmission in the Evolutionary History of Bat Coronaviruses. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2018, 58, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and Evolution of Pathogenic Coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef]
- Olival, K.J.; Hosseini, P.R.; Zambrana-Torrelio, C.; Ross, N.; Bogich, T.L.; Daszak, P. Host and Viral Traits Predict Zoonotic Spillover from Mammals. Nature 2017, 546, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Dhama, K.; Patel, S.K.; Sharun, K.; Pathak, M.; Tiwari, R.; Yatoo, M.I.; Malik, Y.S.; Sah, R.; Rabaan, A.A.; Panwar, P.K.; et al. SARS-CoV-2 Jumping the Species Barrier: Zoonotic Lessons from SARS, MERS and Recent Advances to Combat This Pandemic Virus. Travel Med. Infect. Dis. 2020, 37, 101830. [Google Scholar] [CrossRef] [PubMed]
- Albers, S.; Czech, A. Exploiting TRNAs to Boost Virulence. Life 2016, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Franzo, G.; Tucciarone, C.M.; Cecchinato, M.; Drigo, M. Canine Parvovirus Type 2 (CPV-2) and Feline Panleukopenia Virus (FPV) Codon Bias Analysis Reveals a Progressive Adaptation to the New Niche after the Host Jump. Mol. Phylogenet. Evol. 2017, 114, 82–92. [Google Scholar] [CrossRef]
- Simón, D.; Fajardo, A.; Sóñora, M.; Delfraro, A.; Musto, H. Host Influence in the Genomic Composition of Flaviviruses: A Multivariate Approach. Biochem. Biophys. Res. Commun. 2017, 492, 572–578. [Google Scholar] [CrossRef]
- Rahman, S.U.; Yao, X.; Li, X.; Chen, D.; Tao, S. Analysis of Codon Usage Bias of Crimean-Congo Hemorrhagic Fever Virus and Its Adaptation to Hosts. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2018, 58, 1–16. [Google Scholar] [CrossRef]
- Tian, L.; Shen, X.; Murphy, R.W.; Shen, Y. The Adaptation of Codon Usage of +ssRNA Viruses to Their Hosts. Infect. Genet. Evol. 2018, 63, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Ikemura, T. Codon Usage and TRNA Content in Unicellular and Multicellular Organisms. Mol. Biol. Evol. 1985, 2, 13–34. [Google Scholar] [CrossRef]
- Kanaya, S.; Yamada, Y.; Kinouchi, M.; Kudo, Y.; Ikemura, T. Codon Usage and TRNA Genes in Eukaryotes: Correlation of Codon Usage Diversity with Translation Efficiency and with CG-Dinucleotide Usage as Assessed by Multivariate Analysis. J. Mol. Evol. 2001, 53, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Drummond, D.A.; Wilke, C.O. Mistranslation-Induced Protein Misfolding as a Dominant Constraint on Coding-Sequence Evolution. Cell 2008, 134, 341–352. [Google Scholar] [CrossRef]
- Hershberg, R.; Petrov, D.A. Selection on Codon Bias. Annu. Rev. Genet. 2008, 42, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Dos Reis, M.; Wernisch, L. Estimating Translational Selection in Eukaryotic Genomes. Mol. Biol. Evol. 2009, 26, 451–461. [Google Scholar] [CrossRef]
- Zhou, Z.; Dang, Y.; Zhou, M.; Li, L.; Yu, C.; Fu, J.; Chen, S.; Liu, Y. Codon Usage Is an Important Determinant of Gene Expression Levels Largely through Its Effects on Transcription. Proc. Natl. Acad. Sci. USA 2016, 113, E6117–E6125. [Google Scholar] [CrossRef]
- Kudla, G.; Murray, A.W.; Tollervey, D.; Plotkin, J.B. Coding-Sequence Determinants of Gene Expression in Escherichia Coli. Science 2009, 324, 255–258. [Google Scholar] [CrossRef]
- Goodman, D.B.; Church, G.M.; Kosuri, S. Causes and Effects of N-Terminal Codon Bias in Bacterial Genes. Science 2013, 342, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, M.A.; Kurland, C.G.; Pedersen, S. Codon Usage Determines Translation Rate in Escherichia Coli. J. Mol. Biol. 1989, 207, 365–377. [Google Scholar] [CrossRef]
- Akashi, H. Synonymous Codon Usage in Drosophila Melanogaster: Natural Selection and Translational Accuracy. Genetics 1994, 136, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Presnyak, V.; Alhusaini, N.; Chen, Y.-H.; Martin, S.; Morris, N.; Kline, N.; Olson, S.; Weinberg, D.; Baker, K.E.; Graveley, B.R.; et al. Codon Optimality Is a Major Determinant of MRNA Stability. Cell 2015, 160, 1111–1124. [Google Scholar] [CrossRef]
- Pagani, F.; Raponi, M.; Baralle, F.E. Synonymous Mutations in CFTR Exon 12 Affect Splicing and Are Not Neutral in Evolution. Proc. Natl. Acad. Sci. USA 2005, 102, 6368–6372. [Google Scholar] [CrossRef] [PubMed]
- Bahir, I.; Fromer, M.; Prat, Y.; Linial, M. Viral Adaptation to Host: A Proteome-Based Analysis of Codon Usage and Amino Acid Preferences. Mol. Syst. Biol. 2009, 5, 311. [Google Scholar] [CrossRef]
- Lucks, J.B.; Nelson, D.R.; Kudla, G.R.; Plotkin, J.B. Genome Landscapes and Bacteriophage Codon Usage. PLoS Comput. Biol. 2008, 4, e1000001. [Google Scholar] [CrossRef]
- Wong, E.H.; Smith, D.K.; Rabadan, R.; Peiris, M.; Poon, L.L. Codon Usage Bias and the Evolution of Influenza A Viruses. Codon Usage Biases of Influenza Virus. BMC Evol. Biol. 2010, 10, 253. [Google Scholar] [CrossRef] [PubMed]
- Félez-Sánchez, M.; Trösemeier, J.-H.; Bedhomme, S.; González-Bravo, M.I.; Kamp, C.; Bravo, I.G. Cancer, Warts, or Asymptomatic Infections: Clinical Presentation Matches Codon Usage Preferences in Human Papillomaviruses. Genome Biol. Evol. 2015, 7, 2117–2135. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gu, H.; Chu, D.K.W.; Peiris, M.; Poon, L.L.M. Multivariate Analyses of Codon Usage of SARS-CoV-2 and Other Betacoronaviruses. Virus Evol. 2020, 6, veaa032. [Google Scholar] [CrossRef]
- Tort, F.L.; Castells, M.; Cristina, J. A Comprehensive Analysis of Genome Composition and Codon Usage Patterns of Emerging Coronaviruses. Virus Res. 2020, 283, 197976. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Wen, G.; Jiang, J.; Xie, F. Codon Bias Analysis May Be Insufficient for Identifying Host(s) of a Novel Virus. J. Med. Virol. 2020, 92, 1434–1436. [Google Scholar] [CrossRef]
- Lauring, A.S.; Jones, J.O.; Andino, R. Rationalizing the Development of Live Attenuated Virus Vaccines. Nat. Biotechnol. 2010, 28, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Yap, Y.L.; Zhang, X.W.; Danchin, A. Relationship of SARS-CoV to Other Pathogenic RNA Viruses Explored by Tetranucleotide Usage Profiling. BMC Bioinform. 2003, 4, 43. [Google Scholar] [CrossRef]
- Greenbaum, B.D.; Levine, A.J.; Bhanot, G.; Rabadan, R. Patterns of Evolution and Host Gene Mimicry in Influenza and Other RNA Viruses. PLoS Pathog. 2008, 4, e1000079. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, N.J.; Witteveldt, J.; Evans, D.J.; Simmonds, P. The Influence of CpG and UpA Dinucleotide Frequencies on RNA Virus Replication and Characterization of the Innate Cellular Pathways Underlying Virus Attenuation and Enhanced Replication. Nucleic Acids Res. 2014, 42, 4527–4545. [Google Scholar] [CrossRef] [PubMed]
- Takata, M.A.; Gonçalves-Carneiro, D.; Zang, T.M.; Soll, S.J.; York, A.; Blanco-Melo, D.; Bieniasz, P.D. CG Dinucleotide Suppression Enables Antiviral Defence Targeting Non-Self RNA. Nature 2017, 550, 124–127. [Google Scholar] [CrossRef]
- Tulloch, F.; Atkinson, N.J.; Evans, D.J.; Ryan, M.D.; Simmonds, P. RNA Virus Attenuation by Codon Pair Deoptimisation Is an Artefact of Increases in CpG/UpA Dinucleotide Frequencies. eLife 2014, 3, e04531. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, Y.; Takeuchi, O.; Akira, S. TLR9 as a Key Receptor for the Recognition of DNA. Adv. Drug Deliv. Rev. 2008, 60, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Duret, L. Evolution of Synonymous Codon Usage in Metazoans. Curr. Opin. Genet. Dev. 2002, 12, 640–649. [Google Scholar] [CrossRef]
- Chamary, J.V.; Parmley, J.L.; Hurst, L.D. Hearing Silence: Non-Neutral Evolution at Synonymous Sites in Mammals. Nat. Rev. Genet. 2006, 7, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M. Rate, Molecular Spectrum, and Consequences of Human Mutation. Proc. Natl. Acad. Sci. USA 2010, 107, 961–968. [Google Scholar] [CrossRef]
- Duret, L.; Galtier, N. Biased Gene Conversion and the Evolution of Mammalian Genomic Landscapes. Annu. Rev. Genom. Hum. Genet. 2009, 10, 285–311. [Google Scholar] [CrossRef]
- Hershberg, R.; Petrov, D.A. Evidence That Mutation Is Universally Biased towards AT in Bacteria. PLoS Genet. 2010, 6, e1001115. [Google Scholar] [CrossRef]
- Petrov, D.A.; Hartl, D.L. Patterns of Nucleotide Substitution in Drosophila and Mammalian Genomes. Proc. Natl. Acad. Sci. USA 1999, 96, 1475–1479. [Google Scholar] [CrossRef] [PubMed]
- Haddrill, P.R.; Charlesworth, B. Non-Neutral Processes Drive the Nucleotide Composition of Non-Coding Sequences in Drosophila. Biol. Lett. 2008, 4, 438–441. [Google Scholar] [CrossRef] [PubMed]
- Denver, D.R.; Dolan, P.C.; Wilhelm, L.J.; Sung, W.; Lucas-Lledó, J.I.; Howe, D.K.; Lewis, S.C.; Okamoto, K.; Thomas, W.K.; Lynch, M.; et al. A Genome-Wide View of Caenorhabditis Elegans Base-Substitution Mutation Processes. Proc. Natl. Acad. Sci. USA 2009, 106, 16310–16314. [Google Scholar] [CrossRef]
- Ossowski, S.; Schneeberger, K.; Lucas-Lledó, J.I.; Warthmann, N.; Clark, R.M.; Shaw, R.G.; Weigel, D.; Lynch, M. The Rate and Molecular Spectrum of Spontaneous Mutations in Arabidopsis Thaliana. Science 2010, 327, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.M.; Castillo Morales, A.; Ho, A.T.; Mordstein, C.; Mühlhausen, S.; Watson, S.; Cano, L.; Young, B.; Kudla, G.; Hurst, L.D. Evidence for Strong Mutation Bias toward, and Selection against, U Content in SARS-CoV-2: Implications for Vaccine Design. Mol. Biol. Evol. 2021, 38, 67–83. [Google Scholar] [CrossRef]
- Simmonds, P. Rampant C→U Hypermutation in the Genomes of SARS-CoV-2 and Other Coronaviruses: Causes and Consequences for Their Short- and Long-Term Evolutionary Trajectories. mSphere 2020, 5, e00408-20. [Google Scholar] [CrossRef] [PubMed]
- Vijgen, L.; Keyaerts, E.; Moës, E.; Thoelen, I.; Wollants, E.; Lemey, P.; Vandamme, A.-M.; Ranst, M.V. Complete Genomic Sequence of Human Coronavirus OC43: Molecular Clock Analysis Suggests a Relatively Recent Zoonotic Coronavirus Transmission Event. J. Virol. 2005, 79, 1595–1604. [Google Scholar] [CrossRef] [PubMed]
- Pfefferle, S.; Oppong, S.; Drexler, J.F.; Gloza-Rausch, F.; Ipsen, A.; Seebens, A.; Müller, M.A.; Annan, A.; Vallo, P.; Adu-Sarkodie, Y.; et al. Distant Relatives of Severe Acute Respiratory Syndrome Coronavirus and Close Relatives of Human Coronavirus 229E in Bats, Ghana. Emerg. Infect. Dis. J. 2009, 15, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Huynh, J.; Li, S.; Yount, B.; Smith, A.; Sturges, L.; Olsen, J.C.; Nagel, J.; Johnson, J.B.; Agnihothram, S.; Gates, J.E.; et al. Evidence Supporting a Zoonotic Origin of Human Coronavirus Strain NL63. J. Virol. 2012, 86, 12816–12825. [Google Scholar] [CrossRef]
- Al-Khannaq, M.N.; Ng, K.T.; Oong, X.Y.; Pang, Y.K.; Takebe, Y.; Chook, J.B.; Hanafi, N.S.; Kamarulzaman, A.; Tee, K.K. Molecular Epidemiology and Evolutionary Histories of Human Coronavirus OC43 and HKU1 among Patients with Upper Respiratory Tract Infections in Kuala Lumpur, Malaysia. Virol J. 2016, 13, 33. [Google Scholar] [CrossRef]
- Glémin, S.; Arndt, P.F.; Messer, P.W.; Petrov, D.; Galtier, N.; Duret, L. Quantification of GC-Biased Gene Conversion in the Human Genome. Genome Res. 2015, 25, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Kunec, D.; Osterrieder, N. Codon Pair Bias Is a Direct Consequence of Dinucleotide Bias. Cell Rep. 2016, 14, 55–67. [Google Scholar] [CrossRef]
- Dilucca, M.; Forcelloni, S.; Georgakilas, A.G.; Giansanti, A.; Pavlopoulou, A. Codon Usage and Phenotypic Divergences of SARS-CoV-2 Genes. Viruses 2020, 12, 498. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Wang, W.; Zhao, X.; Zai, J.; Li, X. Cross-Species Transmission of the Newly Identified Coronavirus 2019-NCoV. J. Med. Virol. 2020, 92, 433–440. [Google Scholar] [CrossRef]
- De Maio, N.; Walker, C.R.; Turakhia, Y.; Lanfear, R.; Corbett-Detig, R.; Goldman, N. Mutation Rates and Selection on Synonymous Mutations in SARS-CoV-2. Genome Biol. Evol. 2021, 13, evab087. [Google Scholar] [CrossRef]
- Di Giorgio, S.; Martignano, F.; Torcia, M.G.; Mattiuz, G.; Conticello, S.G. Evidence for Host-Dependent RNA Editing in the Transcriptome of SARS-CoV-2. Sci. Adv. 2020, 6, eabb5813. [Google Scholar] [CrossRef]
- Münk, C.; Willemsen, A.; Bravo, I.G. An Ancient History of Gene Duplications, Fusions and Losses in the Evolution of APOBEC3 Mutators in Mammals. BMC Evol. Biol. 2012, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Anderson, B.D. Evolutionary Paradigms from Ancient and Ongoing Conflicts between the Lentiviral Vif Protein and Mammalian APOBEC3 Enzymes. PLoS Pathog. 2016, 12, e1005958. [Google Scholar] [CrossRef]
- Ito, J.; Gifford, R.J.; Sato, K. Retroviruses Drive the Rapid Evolution of Mammalian APOBEC3 Genes. Proc. Natl. Acad. Sci. USA 2020, 117, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Hayward, J.A.; Tachedjian, M.; Cui, J.; Cheng, A.Z.; Johnson, A.; Baker, M.L.; Harris, R.S.; Wang, L.-F.; Tachedjian, G. Differential Evolution of Antiretroviral Restriction Factors in Pteropid Bats as Revealed by APOBEC3 Gene Complexity. Mol. Biol. Evol. 2018, 35, 1626–1637. [Google Scholar] [CrossRef]
- Jebb, D.; Huang, Z.; Pippel, M.; Hughes, G.M.; Lavrichenko, K.; Devanna, P.; Winkler, S.; Jermiin, L.S.; Skirmuntt, E.C.; Katzourakis, A.; et al. Six Reference-Quality Genomes Reveal Evolution of Bat Adaptations. Nature 2020, 583, 578–584. [Google Scholar] [CrossRef]
- Garcia, E.I.; Emerman, M. Recurrent Loss of APOBEC3H Activity during Primate Evolution. J. Virol. 2018, 92, e00971-18. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Emerman, M.; Malik, H.S.; McLaughlin, R.N. Retrocopying Expands the Functional Repertoire of APOBEC3 Antiviral Proteins in Primates. eLife 2020, 9, e58436. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y.; Yamamoto, K.; Ueda, M.T.; Soper, A.; Konno, Y.; Kimura, I.; Uriu, K.; Kumata, R.; Aso, H.; Misawa, N.; et al. A Role for Gorilla APOBEC3G in Shaping Lentivirus Evolution Including Transmission to Humans. PLoS Pathog. 2020, 16, e1008812. [Google Scholar] [CrossRef]
- Burns, C.C.; Campagnoli, R.; Shaw, J.; Vincent, A.; Jorba, J.; Kew, O. Genetic Inactivation of Poliovirus Infectivity by Increasing the Frequencies of CpG and UpA Dinucleotides within and across Synonymous Capsid Region Codons. J. Virol. 2009, 83, 9957–9969. [Google Scholar] [CrossRef]
- Gaunt, E.; Wise, H.M.; Zhang, H.; Lee, L.N.; Atkinson, N.J.; Nicol, M.Q.; Highton, A.J.; Klenerman, P.; Beard, P.M.; Dutia, B.M.; et al. Elevation of CpG Frequencies in Influenza A Genome Attenuates Pathogenicity but Enhances Host Response to Infection. eLife 2016, 5, e12735. [Google Scholar] [CrossRef]
- Antzin-Anduetza, I.; Mahiet, C.; Granger, L.A.; Odendall, C.; Swanson, C.M. Increasing the CpG Dinucleotide Abundance in the HIV-1 Genomic RNA Inhibits Viral Replication. Retrovirology 2017, 14, 49. [Google Scholar] [CrossRef]
- Ibrahim, A.; Fros, J.; Bertran, A.; Sechan, F.; Odon, V.; Torrance, L.; Kormelink, R.; Simmonds, P. A Functional Investigation of the Suppression of CpG and UpA Dinucleotide Frequencies in Plant RNA Virus Genomes. Sci. Rep. 2019, 9, 18359. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Xia, W.; Baillie, J.K.; McKinnon, K. Modelling Mutational and Selection Pressures on Dinucleotides in Eukaryotic Phyla –Selection against CpG and UpA in Cytoplasmically Expressed RNA and in RNA Viruses. BMC Genom. 2013, 14, 610. [Google Scholar] [CrossRef] [PubMed]
- Fros, J.J.; Dietrich, I.; Alshaikhahmed, K.; Passchier, T.C.; Evans, D.J.; Simmonds, P. CpG and UpA Dinucleotides in Both Coding and Non-Coding Regions of Echovirus 7 Inhibit Replication Initiation Post-Entry. eLife 2017, 6, e29112. [Google Scholar] [CrossRef]
- Cooper, D.A.; Banerjee, S.; Chakrabarti, A.; García-Sastre, A.; Hesselberth, J.R.; Silverman, R.H.; Barton, D.J. RNase L Targets Distinct Sites in Influenza A Virus RNAs. J. Virol. 2015, 89, 2764–2776. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán, R.; Moya, A.; Elena, S.F. The Distribution of Fitness Effects Caused by Single-Nucleotide Substitutions in an RNA Virus. Proc. Natl. Acad. Sci. USA 2004, 101, 8396–8401. [Google Scholar] [CrossRef] [PubMed]
- Peris, J.B.; Davis, P.; Cuevas, J.M.; Nebot, M.R.; Sanjuán, R. Distribution of Fitness Effects Caused by Single-Nucleotide Substitutions in Bacteriophage F1. Genetics 2010, 185, 603–609. [Google Scholar] [CrossRef]
- Jacquier, H.; Birgy, A.; Le Nagard, H.; Mechulam, Y.; Schmitt, E.; Glodt, J.; Bercot, B.; Petit, E.; Poulain, J.; Barnaud, G.; et al. Capturing the Mutational Landscape of the Beta-Lactamase TEM-1. Proc. Natl. Acad. Sci. USA 2013, 110, 13067–13072. [Google Scholar] [CrossRef]
- Fragata, I.; Matuszewski, S.; Schmitz, M.A.; Bataillon, T.; Jensen, J.D.; Bank, C. The Fitness Landscape of the Codon Space across Environments. Heredity 2018, 121, 422–437. [Google Scholar] [CrossRef]
- Williams, M.J.; Zapata, L.; Werner, B.; Barnes, C.P.; Sottoriva, A.; Graham, T.A. Measuring the Distribution of Fitness Effects in Somatic Evolution by Combining Clonal Dynamics with DN/DS Ratios. eLife 2020, 9, e48714. [Google Scholar] [CrossRef]
- Plotkin, J.B.; Kudla, G. Synonymous but Not the Same: The Causes and Consequences of Codon Bias. Nat. Rev. Genet. 2011, 12, 32–42. [Google Scholar] [CrossRef]
- Mordstein, C.; Savisaar, R.; Young, R.S.; Bazile, J.; Talmane, L.; Luft, J.; Liss, M.; Taylor, M.S.; Hurst, L.D.; Kudla, G. Codon Usage and Splicing Jointly Influence MRNA Localization. Cell Syst. 2020, 10, 351–362.e8. [Google Scholar] [CrossRef]
- Galtier, N.; Roux, C.; Rousselle, M.; Romiguier, J.; Figuet, E.; Glémin, S.; Bierne, N.; Duret, L. Codon Usage Bias in Animals: Disentangling the Effects of Natural Selection, Effective Population Size, and GC-Biased Gene Conversion. Mol. Biol. Evol. 2018, 35, 1092–1103. [Google Scholar] [CrossRef]
- Caspersson, T.; Farber, S.; Foley, G.E.; Kudynowski, J.; Modest, E.J.; Simonsson, E.; Wagh, U.; Zech, L. Chemical Differentiation along Metaphase Chromosomes. Exp. Cell Res. 1968, 49, 219–222. [Google Scholar] [CrossRef]
- Holmquist, G.P. Evolution of Chromosome Bands: Molecular Ecology of Noncoding DNA. J. Mol. Evol 1989, 28, 469–486. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.P.C. Codon Usage Bias from TRNA’s Point of View: Redundancy, Specialization, and Efficient Decoding for Translation Optimization. Genome Res. 2004, 14, 2279–2286. [Google Scholar] [CrossRef]
- Martínez, M.A.; Jordan-Paiz, A.; Franco, S.; Nevot, M. Synonymous Virus Genome Recoding as a Tool to Impact Viral Fitness. Trends Microbiol. 2016, 24, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Irigoyen, N.; Firth, A.E.; Jones, J.D.; Chung, B.Y.-W.; Siddell, S.G.; Brierley, I. High-Resolution Analysis of Coronavirus Gene Expression by RNA Sequencing and Ribosome Profiling. PLoS Pathog. 2016, 12, e1005473. [Google Scholar] [CrossRef] [PubMed]
- Ratnakumar, A.; Mousset, S.; Glémin, S.; Berglund, J.; Galtier, N.; Duret, L.; Webster, M.T. Detecting Positive Selection within Genomes: The Problem of Biased Gene Conversion. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 2571–2580. [Google Scholar] [CrossRef]
- Shapiro, M.; Krug, L.T.; MacCarthy, T. Mutational Pressure by Host APOBEC3s More Strongly Affects Genes Expressed Early in the Lytic Phase of Herpes Simplex Virus-1 (HSV-1) and Human Polyomavirus (HPyV) Infection. PLoS Pathog. 2021, 17, e1009560. [Google Scholar] [CrossRef]
- Sola, I.; Almazán, F.; Zúñiga, S.; Enjuanes, L. Continuous and Discontinuous RNA Synthesis in Coronaviruses. Annu. Rev. Virol. 2015, 2, 265–288. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lee, J.-Y.; Yang, J.-S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef] [PubMed]
- Finkel, Y.; Mizrahi, O.; Nachshon, A.; Weingarten-Gabbay, S.; Morgenstern, D.; Yahalom-Ronen, Y.; Tamir, H.; Achdout, H.; Stein, D.; Israeli, O.; et al. The Coding Capacity of SARS-CoV-2. Nature 2021, 589, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Van Dorp, L.; Acman, M.; Richard, D.; Shaw, L.P.; Ford, C.E.; Ormond, L.; Owen, C.J.; Pang, J.; Tan, C.C.S.; Boshier, F.A.T.; et al. Emergence of Genomic Diversity and Recurrent Mutations in SARS-CoV-2. Infect. Genet. Evol. 2020, 83, 104351. [Google Scholar] [CrossRef]
- Matyášek, R.; Kovařík, A. Mutation Patterns of Human SARS-CoV-2 and Bat RaTG13 Coronavirus Genomes Are Strongly Biased Towards C>U Transitions, Indicating Rapid Evolution in Their Hosts. Genes 2020, 11, 761. [Google Scholar] [CrossRef]
- Jenkins, G.M.; Holmes, E.C. The Extent of Codon Usage Bias in Human RNA Viruses and Its Evolutionary Origin. Virus Res. 2003, 92, 1–7. [Google Scholar] [CrossRef]
- Cristina, J.; Moreno, P.; Moratorio, G.; Musto, H. Genome-Wide Analysis of Codon Usage Bias in Ebolavirus. Virus Res. 2015, 196, 87–93. [Google Scholar] [CrossRef]
- Burge, C.; Campbell, A.M.; Karlin, S. Over- and under-Representation of Short Oligonucleotides in DNA Sequences. Proc. Natl. Acad. Sci. USA 1992, 89, 1358–1362. [Google Scholar] [CrossRef]
- Delcher, A.L.; Phillippy, A.; Carlton, J.; Salzberg, S.L. Fast Algorithms for Large-Scale Genome Alignment and Comparison. Nucleic Acids Res. 2002, 30, 2478–2483. [Google Scholar] [CrossRef] [PubMed]
- Mostowy, R.; Croucher, N.J.; Andam, C.P.; Corander, J.; Hanage, W.P.; Marttinen, P. Efficient Inference of Recent and Ancestral Recombination within Bacterial Populations. Mol. Biol. Evol. 2017, 34, 1167–1182. [Google Scholar] [CrossRef] [PubMed]
- Eyre-Walker, A.; Woolfit, M.; Phelps, T. The Distribution of Fitness Effects of New Deleterious Amino Acid Mutations in Humans. Genetics 2006, 173, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Muyle, A.; Serres-Giardi, L.; Ressayre, A.; Escobar, J.; Glémin, S. GC-Biased Gene Conversion and Selection Affect GC Content in the Oryza Genus (Rice). Mol. Biol. Evol. 2011, 28, 2695–2706. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, M.; Lambert, A.; Achaz, G. Accuracy of Demographic Inferences from the Site Frequency Spectrum: The Case of the Yoruba Population. Genetics 2017, 206, 439–449. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daron, J.; Bravo, I.G. Variability in Codon Usage in Coronaviruses Is Mainly Driven by Mutational Bias and Selective Constraints on CpG Dinucleotide. Viruses 2021, 13, 1800. https://doi.org/10.3390/v13091800
Daron J, Bravo IG. Variability in Codon Usage in Coronaviruses Is Mainly Driven by Mutational Bias and Selective Constraints on CpG Dinucleotide. Viruses. 2021; 13(9):1800. https://doi.org/10.3390/v13091800
Chicago/Turabian StyleDaron, Josquin, and Ignacio G. Bravo. 2021. "Variability in Codon Usage in Coronaviruses Is Mainly Driven by Mutational Bias and Selective Constraints on CpG Dinucleotide" Viruses 13, no. 9: 1800. https://doi.org/10.3390/v13091800
APA StyleDaron, J., & Bravo, I. G. (2021). Variability in Codon Usage in Coronaviruses Is Mainly Driven by Mutational Bias and Selective Constraints on CpG Dinucleotide. Viruses, 13(9), 1800. https://doi.org/10.3390/v13091800