
A Journey to the Central Nervous System: Routes of Flaviviral Neuroinvasion in Human Disease



Abstract
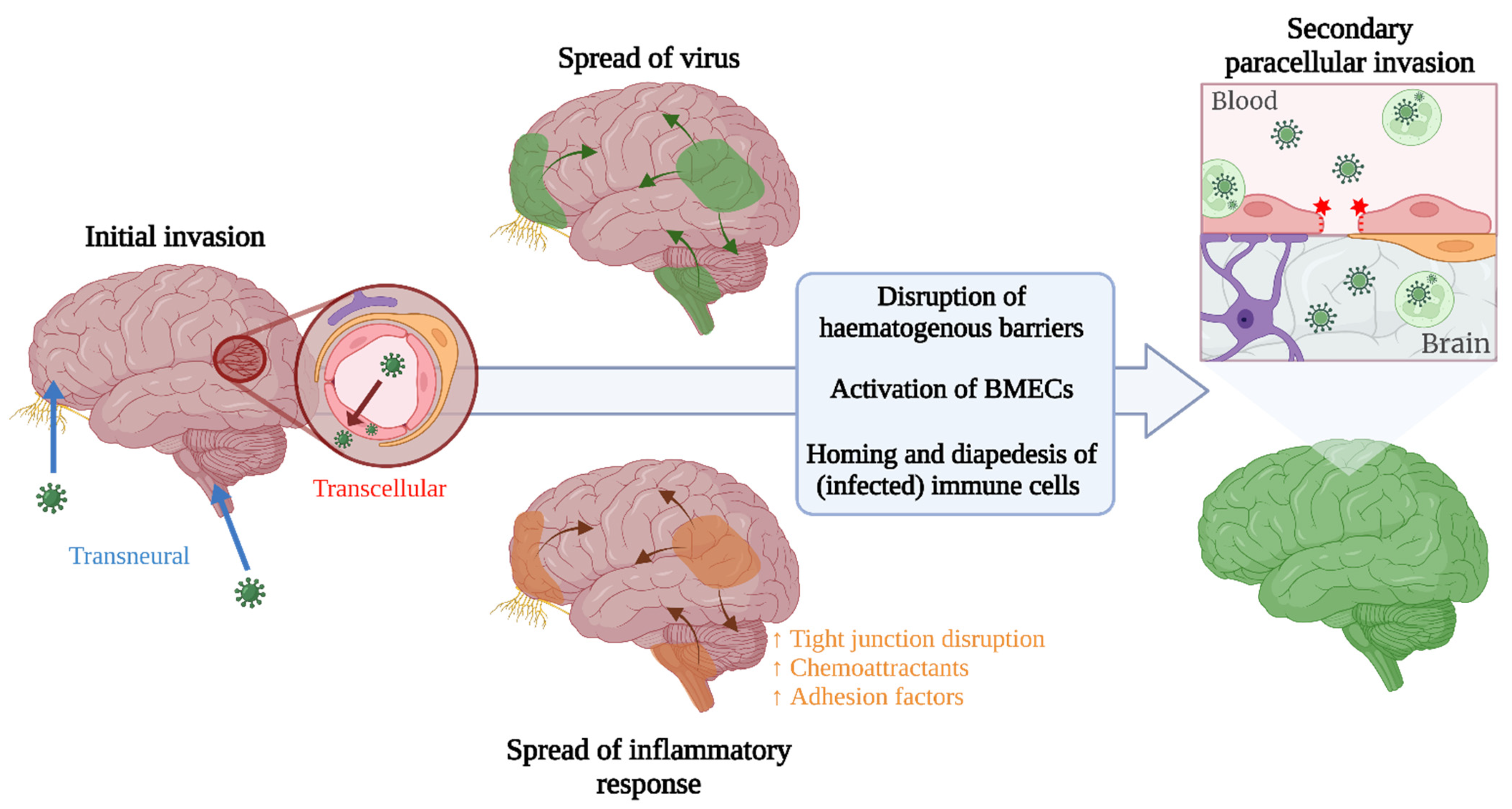
:1. Introduction
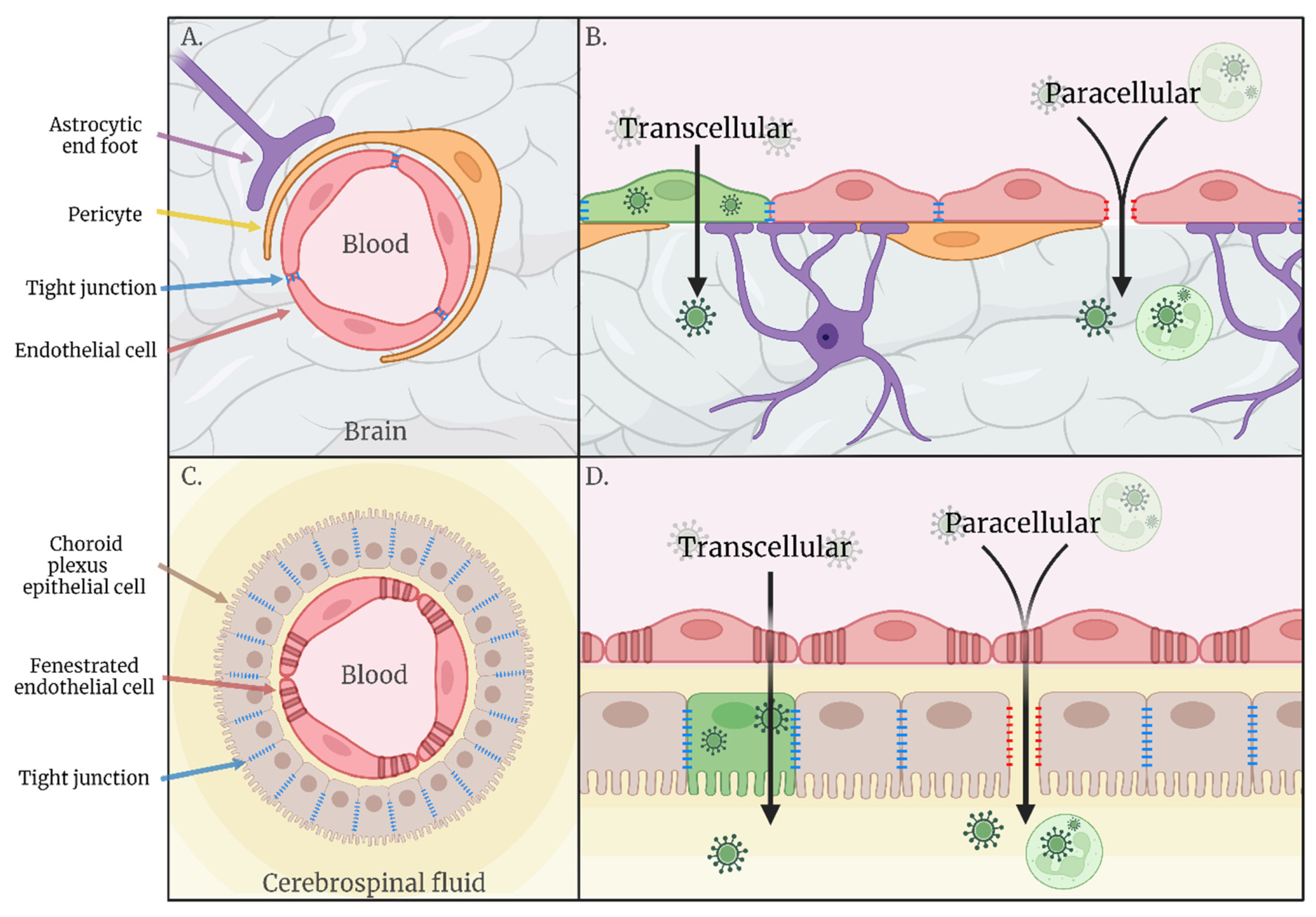
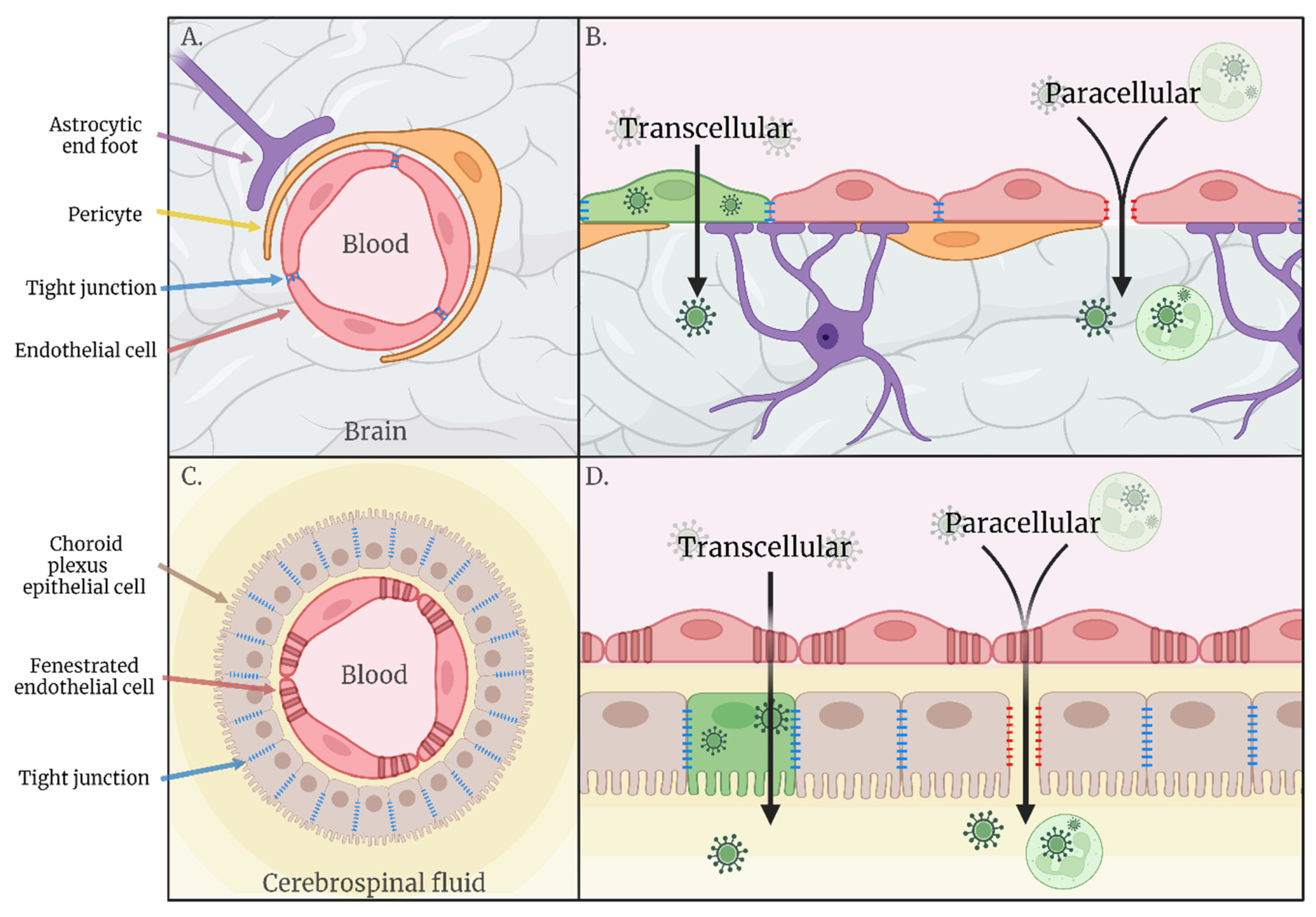
2. Haematogenous Neuroinvasion
2.1. Transcellular
2.2. Paracellular
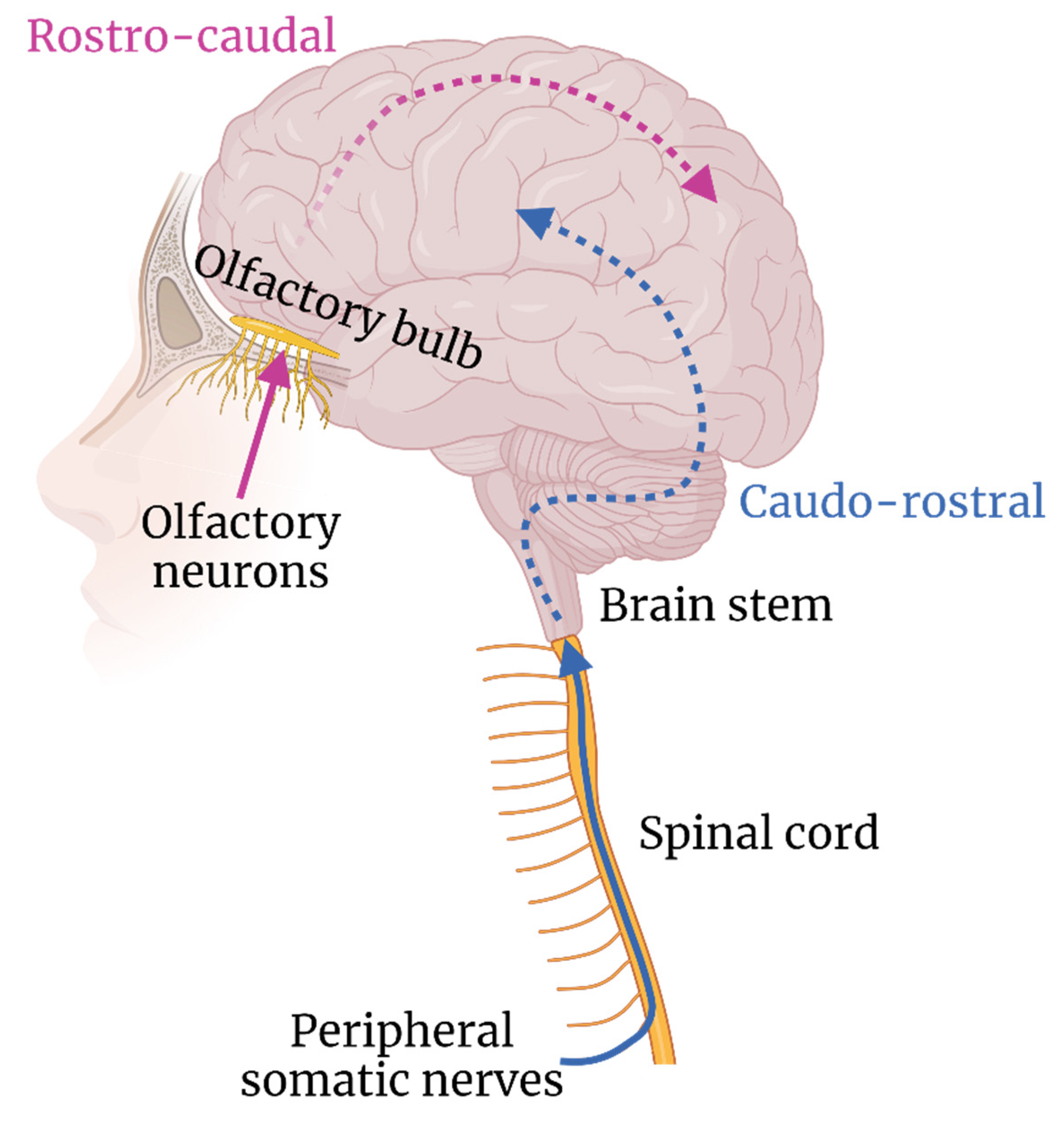
3. Transneural Neuroinvasion
3.1. Peripheral Nerves
3.2. Olfactory Nerves
3.3. Other Possible Routes of Transneural Invasion
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pierson, T.C.; Diamond, M.S. The continued threat of emerging flaviviruses. Nat. Microbiol. 2020, 5, 796–812. [Google Scholar] [CrossRef] [PubMed]
- Al-Obaidi, J.M.M.; Bahadoran, A.; Wang, S.M.; Manikam, R.; Raju, C.S.; Sekaran, S.D. Disruption of the blood brain barrier is vital property of neurotropic viral infection of the central nervous system. Acta Virol. 2018, 62, 16–27. [Google Scholar] [CrossRef]
- Stamatovic, S.M.; Johnson, A.M.; Keep, R.F.; Andjelkovic, A.V. Junctional proteins of the blood-brain barrier: New insights into function and dysfunction. Tissue Barriers 2016, 4, e1154641. [Google Scholar] [CrossRef]
- Jin, J.; Fang, F.; Gao, W.; Chen, H.; Wen, J.; Wen, X.; Chen, J. The Structure and Function of the Glycocalyx and Its Connection with Blood-Brain Barrier. Front. Cell. Neurosci. 2021, 15, 409. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Lo, Y.; Chapagain, M.; Lum, S.; Kumar, M.; Gurjav, U.; Luo, H.; Nakatsuka, A.; Nerurkar, V.R. West Nile virus infection modulates human brain microvascular endothelial cells tight junction proteins and cell adhesion molecules: Transmigration across the in vitro blood-brain barrier. Virology 2009, 385, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.-Y.; Ou, Y.-C.; Chang, C.-Y.; Pan, H.-C.; Chang, C.-J.; Liao, S.-L.; Su, H.-L.; Chen, C.-J. Endothelial Japanese encephalitis virus infection enhances migration and adhesion of leukocytes to brain microvascular endothelia via MEK-dependent expression of ICAM1 and the CINC and RANTES chemokines. J. Neurochem. 2012, 123, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Patabendige, A.; Michael, B.; Craig, A.; Solomon, T. Brain microvascular endothelial-astrocyte cell responses following Japanese encephalitis virus infection in an in vitro human blood-brain barrier model. Mol. Cell. Neurosci. 2018, 89, 60–70. [Google Scholar] [CrossRef]
- Palus, M.; Vancová, M.; Sirmarova, J.; Elsterova, J.; Perner, J.; Ruzek, D. Tick-borne encephalitis virus infects human brain microvascular endothelial cells without compromising blood-brain barrier integrity. Virology 2017, 507, 110–122. [Google Scholar] [CrossRef]
- Hayasaka, D. The Development of Encephalitis Following Tick-Borne Encephalitis Virus Infection in a Mouse Model. In Flavivirus Encephalitis; InTech: Rijeka, Croatia, 2011; pp. 157–166. [Google Scholar]
- Roe, K.; Kumar, M.; Lum, S.; Orillo, B.; Nerurkar, V.R.; Verma, S. West Nile virus-induced disruption of the blood–brain barrier in mice is characterized by the degradation of the junctional complex proteins and increase in multiple matrix metalloproteinases. J. Gen. Virol. 2012, 93, 1193–1203. [Google Scholar] [CrossRef]
- German, A.C.; Myint, K.S.A.; Mai, N.T.H.; Pomeroy, I.; Phu, N.H.; Tzartos, J.; Winter, P.; Collett, J.; Farrar, J.; Barrett, A.; et al. A preliminary neuropathological study of Japanese encephalitis in humans and a mouse model. Trans. R. Soc. Trop. Med. Hyg. 2006, 100, 1135–1145. [Google Scholar] [CrossRef]
- Paddock, C.D.; Nicholson, W.L.; Bhatnagar, J.; Goldsmith, C.S.; Greer, P.W.; Hayes, E.B.; Risko, J.A.; Henderson, C.; Blackmore, C.G.; Lanciotti, R.S.; et al. Fatal Hemorrhagic Fever Caused by West Nile Virus in the United States. Clin. Infect. Dis. 2006, 42, 1527–1535. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chi, X.; Cheng, M.; Huang, X.; Liu, X.; Fan, J.; Xu, H.; Lin, T.; Shi, L.; Qin, C.; et al. Zika virus degrades the ω-3 fatty acid transporter Mfsd2a in brain microvascular endothelial cells and impairs lipid homeostasis. Sci. Adv. 2019, 5, 7142. [Google Scholar] [CrossRef] [PubMed]
- Hasebe, R.; Suzuki, T.; Makino, Y.; Igarashi, M.; Yamanouchi, S.; Maeda, A.; Horiuchi, M.; Sawa, H.; Kimura, T. Transcellular transport of West Nile virus-like particles across human endothelial cells depends on residues 156 and 159 of envelope protein. BMC Microbiol. 2010, 10, 165. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.W.; Nguyen, H.-Y.; Hanna, S.L.; Sánchez, M.D.; Doms, R.W.; Pierson, T.C. West Nile Virus Discriminates between DC-SIGN and DC-SIGNR for Cellular Attachment and Infection. J. Virol. 2006, 80, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- McMinn, P.C.; Lee, E.; Hartley, S.; Roehrig, J.T.; Dalgarno, L.; Weir, R.O. Murray Valley Encephalitis Virus Envelope Protein Antigenic Variants with Altered Hemagglutination Properties and Reduced Neuroinvasiveness in Mice. Virology 1995, 211, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Lobigs, M. Mechanism of Virulence Attenuation of Glycosaminoglycan-Binding Variants of Japanese Encephalitis Virus and Murray Valley Encephalitis Virus. J. Virol. 2002, 76, 4901–4911. [Google Scholar] [CrossRef]
- Bernard, K.A.; Klimstra, W.B.; Johnston, R.E. Mutations in the E2 Glycoprotein of Venezuelan Equine Encephalitis Virus Confer Heparan Sulfate Interaction, Low Morbidity, and Rapid Clearance from Blood of Mice. Virology 2000, 276, 93–103. [Google Scholar] [CrossRef]
- Byrnes, A.P.; Griffin, D.E. Large-Plaque Mutants of Sindbis Virus Show Reduced Binding to Heparan Sulfate, Heightened Viremia, and Slower Clearance from the Circulation. J. Virol. 2000, 74, 644–651. [Google Scholar] [CrossRef]
- Gardner, C.L.; Ebel, G.D.; Ryman, K.D.; Klimstra, W.B. Heparan sulfate binding by natural eastern equine encephalitis viruses promotes neurovirulence. Proc. Natl. Acad. Sci. USA 2011, 108, 16026–16031. [Google Scholar] [CrossRef]
- de Boer, S.M.; Kortekaas, J.; de Haan, C.A.M.; Rottier, P.J.M.; Moormann, R.J.M.; Bosch, B.J. Heparan Sulfate Facilitates Rift Valley Fever Virus Entry into the Cell. J. Virol. 2012, 86, 13767–13771. [Google Scholar] [CrossRef] [Green Version]
- Nybakken, G.E.; Nelson, C.A.; Chen, B.R.; Diamond, M.S.; Fremont, D.H. Crystal Structure of the West Nile Virus Envelope Glycoprotein. J. Virol. 2006, 80, 11467–11474. [Google Scholar] [CrossRef] [PubMed]
- Clé, M.; Barthelemy, J.; Desmetz, C.; Foulongne, V.; Lapeyre, L.; Bolloré, K.; Tuaillon, E.; Erkilic, N.; Kalatzis, V.; Lecollinet, S.; et al. Study of Usutu virus neuropathogenicity in mice and human cellular models. PLoS Negl. Trop. Dis. 2020, 14, e0008223. [Google Scholar] [CrossRef] [PubMed]
- Clé, M.; Constant, O.; Barthelemy, J.; Desmetz, C.; Martin, M.F.; Lapeyre, L.; Cadar, D.; Savini, G.; Teodori, L.; Monaco, F.; et al. Differential neurovirulence of Usutu virus lineages in mice and neuronal cells. J. Neuroinflamm. 2021, 18, 11. [Google Scholar] [CrossRef] [PubMed]
- Weissenböck, H.; Bakonyi, T.; Chvala, S.; Nowotny, N. Experimental Usutu virus infection of suckling mice causes neuronal and glial cell apoptosis and demyelination. Acta Neuropathol. 2004, 108, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, S.A.; Lu, L.; Xiao, S.-Y.; Da Rosa, A.P.A.T.; Tesh, R.B. An Animal Model for Studying the Pathogenesis of Chikungunya Virus Infection. Am. J. Trop. Med. Hyg. 2008, 79, 133–139. [Google Scholar] [CrossRef]
- Kim, J.; Alejandro, B.; Hetman, M.; Hattab, E.M.; Joiner, J.; Schroten, H.; Ishikawa, H.; Chung, D.-H. Zika virus infects pericytes in the choroid plexus and enters the central nervous system through the blood-cerebrospinal fluid barrier. PLoS Pathog. 2020, 16, 841437. [Google Scholar] [CrossRef]
- Hunsperger, E.A.; Roehrig, J.T. Nocodazole delays viral entry into the brain following footpad inoculation with West Nile virus in mice. J. Neurovirol. 2009, 15, 211–218. [Google Scholar] [CrossRef]
- Ricklin, M.E.; Garcìa-Nicolàs, O.; Brechbühl, D.; Python, S.; Zumkehr, B.; Posthaus, H.; Oevermann, A.; Summerfield, A. Japanese encephalitis virus tropism in experimentally infected pigs. Vet. Res. 2016, 47, 34. [Google Scholar] [CrossRef]
- Correale, J.; Villa, A. Cellular Elements of the Blood-Brain Barrier. Neurochem. Res. 2009, 34, 2067–2077. [Google Scholar] [CrossRef]
- Agrawal, T.; Sharvani, V.; Nair, D.; Medigeshi, G.R. Japanese Encephalitis Virus Disrupts Cell-Cell Junctions and Affects the Epithelial Permeability Barrier Functions. PLoS ONE 2013, 8, e69465. [Google Scholar] [CrossRef] [Green Version]
- Meertens, L.; Labeau, A.; Dejarnac, O.; Cipriani, S.; Sinigaglia, L.; Bonnet-Madin, L.; Le Charpentier, T.; Hafirassou, M.L.; Zamborlini, A.; Cao-Lormeau, V.-M.; et al. Axl Mediates ZIKA Virus Entry in Human Glial Cells and Modulates Innate Immune Responses. Cell Rep. 2017, 18, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Meertens, L.; Carnec, X.; Lecoin, M.P.; Ramdasi, R.; Guivel-Benhassine, F.; Lew, E.; Lemke, G.; Schwartz, O.; Amara, A. The TIM and TAM Families of Phosphatidylserine Receptors Mediate Dengue Virus Entry. Cell Host Microbe 2012, 12, 544–557. [Google Scholar] [CrossRef] [PubMed]
- Hastings, A.K.; Hastings, K.; Uraki, R.; Hwang, J.; Gaitsch, H.; Dhaliwal, K.; Williamson, E.; Fikrig, E. Loss of the TAM Receptor Axl Ameliorates Severe Zika Virus Pathogenesis and Reduces Apoptosis in Microglia. iScience 2019, 13, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Miner, J.J.; Daniels, B.P.; Shrestha, B.; Proenca-Modena, J.L.; Lew, E.D.; Lazear, H.; Gorman, M.J.; Lemke, G.; Klein, R.S.; Diamond, M.S. The TAM receptor Mertk protects against neuroinvasive viral infection by maintaining blood-brain barrier integrity. Nat. Med. 2015, 21, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-Y.; Zhen, Z.-D.; Fan, D.-Y.; Wang, P.-G.; An, J. Axl Alleviates Neuroinflammation and Delays Japanese Encephalitis Progression in Mice. Virol. Sin. 2021, 36, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.J.; Antonetti, D.A. The role of small GTPases and EPAC-Rap signaling in the regulation of the blood-brain and blood-retinal barriers. Tissue Barriers 2017, 5, e1339768. [Google Scholar] [CrossRef]
- Daniels, B.P.; Holman, D.W.; Cruz-Orengo, L.; Jujjavarapu, H.; Durrant, D.M.; Klein, R.S. Viral Pathogen-Associated Molecular Patterns Regulate Blood-Brain Barrier Integrity via Competing Innate Cytokine Signals. mBio 2014, 5, e01476-14. [Google Scholar] [CrossRef]
- Lazear, H.M.; Daniels, B.P.; Pinto, A.K.; Huang, A.C.; Vick, S.C.; Doyle, S.E.; Gale, M.; Klein, R.S.; and Diamond, M.S. Interferon-λ restricts West Nile virus neuroinvasion by tightening the blood-brain barrier. Sci. Transl. Med. 2015, 7, 284ra57. [Google Scholar] [CrossRef]
- Yang, C.-M.; Lin, C.-C.; Lee, I.-T.; Lin, Y.-H.; Yang, C.M.; Chen, W.-J.; Jou, M.-J.; Hsiao, L.-D. Japanese encephalitis virus induces matrix metalloproteinase-9 expression via a ROS/c-Src/PDGFR/PI3K/Akt/MAPKs-dependent AP-1 pathway in rat brain astrocytes. J. Neuroinflamm. 2012, 9, 517. [Google Scholar] [CrossRef]
- Palus, M.; Bílý, T.; Elsterová, J.; Langhansová, H.; Salát, J.; Vancová, M.; Růžek, D. Infection and injury of human astrocytes by tick-borne encephalitis virus. J. Gen. Virol. 2014, 95, 2411–2426. [Google Scholar] [CrossRef]
- Verma, S.; Kumar, M.; Gurjav, U.; Lum, S.; Nerurkar, V.R. Reversal of West Nile virus-induced blood–brain barrier disruption and tight junction proteins degradation by matrix metalloproteinases inhibitor. Virology 2010, 397, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Dai, J.; Bai, F.; Kong, K.-F.; Wong, S.J.; Montgomery, R.R.; Madri, J.A.; Fikrig, E. Matrix Metalloproteinase 9 Facilitates West Nile Virus Entry into the Brain. J. Virol. 2008, 82, 8978–8985. [Google Scholar] [CrossRef] [PubMed]
- Kanoh, Y.; Ohara, T.; Kanoh, M.; Akahoshi, T. Serum Matrix Metalloproteinase-2 Levels Indicate Blood–CSF Barrier Damage in Patients with Infectious Meningitis. Inflammation 2008, 31, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Vandenbroucke, R.; Dejonckheere, E.; Van Lint, P.; Demeestere, D.; Van Wonterghem, E.; Vanlaere, I.; Puimège, L.; Van Hauwermeiren, F.; De Rycke, R.; Mc Guire, C.; et al. Matrix Metalloprotease 8-Dependent Extracellular Matrix Cleavage at the Blood-CSF Barrier Contributes to Lethality during Systemic Inflammatory Diseases. J. Neurosci. 2012, 32, 9805–9816. [Google Scholar] [CrossRef]
- Chen, S.-T.; Liu, R.-S.; Wu, M.-F.; Lin, Y.-L.; Chen, S.-Y.; Tan, D.T.-W.; Chou, T.-Y.; Tsai, I.-S.; Li, L.; Hsieh, S.-L. CLEC5A Regulates Japanese Encephalitis Virus-Induced Neuroinflammation and Lethality. PLoS Pathog. 2012, 8, e1002655. [Google Scholar] [CrossRef]
- Haruwaka, K.; Ikegami, A.; Tachibana, Y.; Ohno, N.; Konishi, H.; Hashimoto, A.; Matsumoto, M.; Kato, D.; Ono, R.; Kiyama, H.; et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun. 2019, 10, 5816. [Google Scholar] [CrossRef]
- Varatharaj, A.; Galea, I. The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 2017, 60, 1–12. [Google Scholar] [CrossRef]
- Skelly, D.T.; Hennessy, E.; Dansereau, M.A.; Cunningham, C. A Systematic Analysis of the Peripheral and CNS Effects of Systemic LPS, IL-1Β, TNF-α and IL-6 Challenges in C57BL/6 Mice. PLoS ONE 2013, 8, e69123. [Google Scholar] [CrossRef]
- Winter, P.M.; Dung, N.M.; Loan, H.T.; Kneen, R.; Wills, B.; Thu, L.T.; House, D.; White, N.J.; Farrar, J.; Hart, C.A.; et al. Proinflammatory Cytokines and Chemokines in Humans with Japanese Encephalitis. J. Infect. Dis. 2004, 190, 1618–1626. [Google Scholar] [CrossRef]
- Almeida, R.S.; Ferreira, M.L.B.; Sonon, P.; Cordeiro, M.T.; Sadissou, I.; Diniz, G.T.N.; Militão-Albuquerque, M.D.F.P.; Franca, R.F.D.O.; Donadi, E.A.; Lucena-Silva, N. Cytokines and Soluble HLA-G Levels in the Acute and Recovery Phases of Arbovirus-Infected Brazilian Patients Exhibiting Neurological Complications. Front. Immunol. 2021, 12, 582935. [Google Scholar] [CrossRef]
- Dupuis-Maguiraga, L.; Noret, M.; Brun, S.; Le Grand, R.; Gras, G.; Roques, P. Chikungunya Disease: Infection-Associated Markers from the Acute to the Chronic Phase of Arbovirus-Induced Arthralgia. PLoS Negl. Trop. Dis. 2012, 6, e1446. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.; Thakar, J.; Yuan, X.; Nolan, M.; Murray, K.O.; Lee, W.T.; Wong, S.J.; Meng, H.; Fikrig, E.; Kleinstein, S.H.; et al. Immune Markers Associated with Host Susceptibility to Infection with West Nile Virus. Viral Immunol. 2014, 27, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Li, Y.; Wei, J.; Zhang, Y.; Bian, C.; Wang, K.; Wu, X.; Hu, Y.; Li, J.; Yang, Y. Elevation of Matrix Metalloproteinase-9 Level in Cerebrospinal Fluid of Tick-Borne Encephalitis Patients Is Associated with IgG Extravassation and Disease Severity. PLoS ONE 2013, 8, e77427. [Google Scholar] [CrossRef]
- Palus, M.; Žampachová, E.; Elsterová, J.; Růžek, D. Serum matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 levels in patients with tick-borne encephalitis. J. Infect. 2014, 68, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Constant, O.; Barthelemy, J.; Nagy, A.; Salinas, S.; Simonin, Y. West Nile Virus Neuroinfection in Humans: Peripheral Biomarkers of Neuroinflammation and Neuronal Damage. Viruses 2022, 14, 756. [Google Scholar] [CrossRef] [PubMed]
- Ramos, H.J.; Lanteri, M.C.; Blahnik, G.; Negash, A.; Suthar, M.S.; Brassil, M.M.; Sodhi, K.; Treuting, P.M.; Busch, M.P.; Norris, P.J.; et al. IL-1β Signaling Promotes CNS-Intrinsic Immune Control of West Nile Virus Infection. PLoS Pathog. 2012, 8, e1003039. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, P.; Kindsvogel, W.; Xu, W.; Henderson, K.; Schlutsmeyer, S.; Whitmore, T.E.; Kuestner, R.; Garrigues, U.; Birks, C.; Roraback, J.; et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 2003, 4, 63–68. [Google Scholar] [CrossRef]
- Wang, Y.; Jin, S.; Sonobe, Y.; Cheng, Y.; Horiuchi, H.; Parajuli, B.; Kawanokuchi, J.; Mizuno, T.; Takeuchi, H.; Suzumura, A. Interleukin-1β Induces Blood–Brain Barrier Disruption by Downregulating Sonic Hedgehog in Astrocytes. PLoS ONE 2014, 9, e110024. [Google Scholar] [CrossRef]
- Wang, T.; Town, T.; Alexopoulou, L.; Anderson, J.F.; Fikrig, E.; Flavell, R.A. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat. Med. 2004, 10, 1366–1373. [Google Scholar] [CrossRef]
- Puerta-Guardo, H.; Glasner, D.R.; Espinosa, D.A.; Biering, S.B.; Patana, M.; Ratnasiri, K.; Wang, C.; Beatty, P.R.; Harris, E. Flavivirus NS1 Triggers Tissue-Specific Vascular Endothelial Dysfunction Reflecting Disease Tropism. Cell Rep. 2019, 26, 1598. [Google Scholar] [CrossRef] [Green Version]
- Puerta-Guardo, H.; Glasner, D.; Harris, E. Dengue Virus NS1 Disrupts the Endothelial Glycocalyx, Leading to Hyperpermeability. PLoS Pathog. 2016, 12, e1005738. [Google Scholar] [CrossRef] [PubMed]
- Modhiran, N.; Watterson, D.; Muller, D.A.; Panetta, A.K.; Sester, D.P.; Liu, L.; Hume, D.A.; Stacey, K.J.; Young, P.R. Dengue virus NS1 protein activates cells via Toll-like receptor 4 and disrupts endothelial cell monolayer integrity. Sci. Transl. Med. 2015, 7, 304ra142. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, S.; Chakravarty, A. Neurological Complications of Dengue Fever. Curr. Neurol. Neurosci. Rep. 2022, 22, 515–529. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Qi, J.; Haywood, J.; Shi, Y.; Gao, G.F. Zika virus NS1 structure reveals diversity of electrostatic surfaces among flaviviruses. Nat. Struct. Mol. Biol. 2016, 23, 456–458. [Google Scholar] [CrossRef]
- Libraty, D.H.; Young, P.; Pickering, D.; Endy, T.P.; Kalayanarooj, S.; Green, S.; Vaughn, D.W.; Nisalak, A.; Ennis, F.A.; Rothman, A. High Circulating Levels of the Dengue Virus Nonstructural Protein NS1 Early in Dengue Illness Correlate with the Development of Dengue Hemorrhagic Fever. J. Infect. Dis. 2002, 186, 1165–1168. [Google Scholar] [CrossRef]
- Young, P.R.; Hilditch, P.A.; Bletchly, C.; Halloran, W. An Antigen Capture Enzyme-Linked Immunosorbent Assay Reveals High Levels of the Dengue Virus Protein NS1 in the Sera of Infected Patients. J. Clin. Microbiol. 2000, 38, 1053–1057. [Google Scholar] [CrossRef]
- Shen, J.; T-To, S.S.; Schrieber, L.; King, N.J. Early E-selectin, VCAM-1, ICAM-1, and late major histocompatibility complex antigen induction on human endothelial cells by flavivirus and comodulation of adhesion molecule expression by immune cytokines. J. Virol. 1997, 71, 9323–9332. [Google Scholar] [CrossRef]
- Garcia-Tapia, D.; Hassett, D.E.; Mitchell, W.J.; Johnson, G.C.; Kleiboeker, S.B. West Nile virus encephalitis: Sequential histopathological and immunological events in a murine model of infection. J. Neurovirol. 2007, 13, 130–138. [Google Scholar] [CrossRef]
- Andrews, D.M.; Matthews, V.B.; Sammels, L.M.; Carrello, A.C.; McMinn, P.C. The Severity of Murray Valley Encephalitis in Mice Is Linked to Neutrophil Infiltration and Inducible Nitric Oxide Synthase Activity in the Central Nervous System. J. Virol. 1999, 73, 8781–8790. [Google Scholar] [CrossRef]
- Dai, J.; Wang, P.; Bai, F.; Town, T.; Fikrig, E. ICAM-1 Participates in the Entry of West Nile Virus into the Central Nervous System. J. Virol. 2008, 82, 4164. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Roe, K.; Nerurkar, P.V.; Orillo, B.; Thompson, K.S.; Verma, S.; Nerurkar, V.R. Reduced immune cell infiltration and increased pro-inflammatory mediators in the brain of Type 2 diabetic mouse model infected with West Nile virus. J. Neuroinflamm. 2014, 11, 80. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Welte, T.; McGargill, M.; Town, T.; Thompson, J.; Anderson, J.F.; Flavell, R.A.; Fikrig, E.; Hedrick, S.; Wang, T. Drak2 Contributes to West Nile Virus Entry into the Brain and Lethal Encephalitis. J. Immunol. 2008, 181, 2084–2091. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Hasegawa, Y.; Kanamaru, K.; Zhang, J.H. Mechanisms of Osteopontin-Induced Stabilization of Blood-Brain Barrier Disruption after Subarachnoid Hemorrhage in Rats. Stroke 2010, 41, 1783–1790. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Acharya, D.; Duty, L.; Thompson, E.A.; Le, L.; Stokic, D.S.; Leis, A.A.; Bai, F. Osteopontin facilitates West Nile virus neuroinvasion via neutrophil “Trojan horse” transport. Sci. Rep. 2017, 7, 4722. [Google Scholar] [CrossRef]
- Wang, Z.-Y.; Zhen, Z.-D.; Fan, D.-Y.; Qin, C.-F.; Han, D.-S.; Zhou, H.-N.; Wang, P.-G.; An, J. Axl Deficiency Promotes the Neuroinvasion of Japanese Encephalitis Virus by Enhancing IL-1α Production from Pyroptotic Macrophages. J. Virol. 2020, 94, 602–622. [Google Scholar] [CrossRef]
- Ben-Zvi, A.; Lacoste, B.; Kur, E.; Andreone, B.J.; Mayshar, Y.; Yan, H.; Gu, C. Mfsd2a is critical for the formation and function of the blood–brain barrier. Nature 2014, 509, 507. [Google Scholar] [CrossRef]
- DeBiasi, R.L.; Tyler, K.L. West Nile virus meningoencephalitis. Nat. Clin. Pract. Neurol. 2006, 2, 264–275. [Google Scholar] [CrossRef]
- Sejvar, J.J.; Bode, A.V.; Marfin, A.A.; Campbell, G.L.; Ewing, D.; Mazowiecki, M.; Pavot, P.V.; Schmitt, J.; Pape, J.; Biggerstaff, B.J.; et al. West Nile Virus–associated Flaccid Paralysis. Emerg. Infect. Dis. 2005, 11, 1021–1027. [Google Scholar] [CrossRef]
- Samuel, M.A.; Wang, H.; Siddharthan, V.; Morrey, J.D.; Diamond, M.S. Axonal transport mediates West Nile virus entry into the central nervous system and induces acute flaccid paralysis. Proc. Natl. Acad. Sci. USA 2007, 104, 17140–17145. [Google Scholar] [CrossRef]
- Wang, H.; Siddharthan, V.; Hall, J.O.; Morrey, J.D. West Nile virus preferentially transports along motor neuron axons after sciatic nerve injection of hamsters. J. Neurovirol. 2009, 15, 293–299. [Google Scholar] [CrossRef]
- Misra, U.K.; Kalita, J. Anterior horn cells are also involved in Japanese encephalitis. Acta Neurol. Scand. 2009, 96, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Solomon, T.; Kneen, R.; Dung, N.M.; Khanh, V.C.; Thuy, T.T.N.; Ha, D.Q.; Day, N.P.; Nisalak, A.; Vaughn, D.W.; White, N.J. Poliomyelitis-like illness due to Japanese encephalitis virus. Lancet 1998, 351, 1094–1097. [Google Scholar] [CrossRef]
- Douglas, M.W.; Stephens, D.P.; Burrow, J.N.; Anstey, N.M.; Talbot, K.; Currie, B.J. Murray Valley encephalitis in an adult traveller complicated by long-term flaccid paralysis: Case report and review of the literature. Trans. R. Soc. Trop. Med. Hyg. 2007, 101, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, L.; Vapalahti, O. Tick-borne encephalitis. Lancet 2008, 371, 1861–1871. [Google Scholar] [CrossRef]
- Beer, S.; Brune, N.; Kesselring, J. Detection of anterior horn lesions by MRI in central European tick-borne encephalomyelitis. J. Neurol. 1999, 246, 1169–1171. [Google Scholar] [CrossRef] [PubMed]
- Douglas, M.W.; Diefenbach, R.J.; Homa, F.L.; Miranda-Saksena, M.; Rixon, F.J.; Vittone, V.; Byth, K.; Cunningham, A.L. Herpes Simplex Virus Type 1 Capsid Protein VP26 Interacts with Dynein Light Chains RP3 and Tctex1 and Plays a Role in Retrograde Cellular Transport. J. Biol. Chem. 2004, 279, 28522–28530. [Google Scholar] [CrossRef]
- Mueller, S.; Cao, X.; Welker, R.; Wimmer, E. Interaction of the Poliovirus Receptor CD155 with the Dynein Light Chain Tctex-1 and Its Implication for Poliovirus Pathogenesis. J. Biol. Chem. 2002, 277, 7897–7904. [Google Scholar] [CrossRef]
- Brault, J.-B.; Kudelko, M.; Vidalain, P.-O.; Tangy, F.; Desprès, P.; Pardigon, N. The interaction of flavivirus M protein with light chain Tctex-1 of human dynein plays a role in late stages of virus replication. Virology 2011, 417, 369–378. [Google Scholar] [CrossRef]
- Durrant, D.M.; Ghosh, S.; Klein, R.S. The Olfactory Bulb: An Immunosensory Effector Organ during Neurotropic Viral Infections. ACS Chem. Neurosci. 2016, 7, 464–469. [Google Scholar] [CrossRef]
- Nir, Y.; Beemer, A.; Goldwasser, R.A. West Nile Virus infection in mice following exposure to a viral aerosol. Br. J. Exp. Pathol. 1965, 46, 443–449. [Google Scholar]
- Brown, A.N.; Kent, K.A.; Bennett, C.J.; Bernard, K.A. Tissue tropism and neuroinvasion of West Nile virus do not differ for two mouse strains with different survival rates. Virology 2007, 368, 422–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Riel, D.; Verdijk, R.M.; Kuiken, T. The olfactory nerve: A shortcut for influenza and other viral diseases into the central nervous system. J. Pathol. 2014, 235, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Impoinvil, D.E.; Baylis, M.; Solomon, T. Japanese Encephalitis: On the One Health Agenda; Springer: Berlin/Heidelberg, Germany, 2012; pp. 205–247. [Google Scholar] [CrossRef]
- van den Hurk, A.F.; Ritchie, S.A.; Mackenzie, J.S. Ecology and Geographical Expansion of Japanese Encephalitis Virus. Annu. Rev. Èntomol. 2009, 54, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, M.E.; García-Nicolás, O.; Brechbühl, D.; Python, S.; Zumkehr, B.; Nougairede, A.; Charrel, R.N.; Posthaus, H.; Oevermann, A.; Summerfield, A. Vector-free transmission and persistence of Japanese encephalitis virus in pigs. Nat. Commun. 2016, 7, 10832. [Google Scholar] [CrossRef]
- Yamada, M.; Nakamura, K.; Yoshii, M.; Kaku, Y.; Narita, M. Brain Lesions Induced by Experimental Intranasal Infection of Japanese Encephalitis Virus in Piglets. J. Comp. Pathol. 2009, 141, 156–162. [Google Scholar] [CrossRef]
- Park, S.L.; Huang, Y.-J.S.; Lyons, A.C.; Ayers, V.B.; Hettenbach, S.M.; McVey, D.S.; Burton, K.R.; Higgs, S.; VanLandingham, D.L. North American domestic pigs are susceptible to experimental infection with Japanese encephalitis virus. Sci. Rep. 2018, 8, 7951. [Google Scholar] [CrossRef]
- McMINN, P.C.; Dalgarno, L.; Weir, R.C. A Comparison of the Spread of Murray Valley Encephalitis Viruses of High or Low Neuroinvasiveness in the Tissues of Swiss Mice after Peripheral Inoculation. Virology 1996, 220, 414–423. [Google Scholar] [CrossRef]
- Niven, D.J.; Afra, K.; Iftinca, M.; Tellier, R.; Fonseca, K.; Kramer, A.; Safronetz, D.; Holloway, K.; Drebot, M.; Johnson, A.S. Fatal Infection with Murray Valley Encephalitis Virus Imported from Australia to Canada, 2011. Emerg. Infect. Dis. 2017, 23, 280–283. [Google Scholar] [CrossRef]
- Speers, D.J.; Flexman, J.; Blyth, C.C.; Rooban, N.; Raby, E.; Ramaseshan, G.; Benson, S.; Smith, D.W. Clinical and Radiological Predictors of Outcome for Murray Valley Encephalitis. Am. J. Trop. Med. Hyg. 2013, 88, 481–489. [Google Scholar] [CrossRef]
- Knox, J.; Cowan, R.U.; Doyle, J.S.; Ligtermoet, M.K.; Archer, J.S.; Burrow, J.N.C.; Tong, S.; Currie, B.J.; MacKenzie, J.S.; Smith, D.W.; et al. Murray Valley encephalitis: A review of clinical features, diagnosis and treatment. Med. J. Aust. 2012, 196, 322–326. [Google Scholar] [CrossRef] [Green Version]
- Balogh, Z.; Ferenczi, E.; Szeles, K.; Stefanoff, P.; Gut, W.; Szomor, K.N.; Takacs, M.; Berencsi, G. Tick-borne encephalitis outbreak in Hungary due to consumption of raw goat milk. J. Virol. Methods 2010, 163, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Hudopisk, N.; Korva, M.; Janet, E.; Simetinger, M.; Grgič-Vitek, M.; Gubensek, J.; Natek, V.; Kraigher, A.; Strle, F.; Avšič-Županc, T. Tick-borne Encephalitis Associated with Consumption of Raw Goat Milk, Slovenia, 2012. Emerg. Infect. Dis. 2013, 19, 806–808. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Achazi, K.; Moller, L.; Schulzke, J.D.; Niedrig, M.; Bücker, R. Tick-Borne Encephalitis Virus Replication, Intracellular Trafficking, and Pathogenicity in Human Intestinal Caco-2 Cell Monolayers. PLoS ONE 2014, 9, e96957. [Google Scholar] [CrossRef] [PubMed]
- Kaelberer, M.M.; Buchanan, K.L.; Klein, M.E.; Barth, B.B.; Montoya, M.M.; Shen, X.; Bohórquez, D.V. A gut-brain neural circuit for nutrient sensory transduction. Science 2018, 361, eaat5236. [Google Scholar] [CrossRef]
- White, J.P.; Xiong, S.; Malvin, N.P.; Khoury-Hanold, W.; Heuckeroth, R.O.; Stappenbeck, T.S.; Diamond, M.S. Intestinal Dysmotility Syndromes following Systemic Infection by Flaviviruses. Cell 2018, 175, 1198–1212.e12. [Google Scholar] [CrossRef]
- Sejvar, J.J. Clinical Manifestations and Outcomes of West Nile Virus Infection. Viruses 2014, 6, 606–623. [Google Scholar] [CrossRef]
- Wojaczynski, G.J.; Engel, E.; Steren, K.E.; Enquist, L.W.; Card, J.P. The neuroinvasive profiles of H129 (herpes simplex virus type 1) recombinants with putative anterograde-only transneuronal spread properties. Anat. Embryol. 2014, 220, 1395–1420. [Google Scholar] [CrossRef]
- Brittle, E.E.; Reynolds, A.E.; Enquist, L.W. Two Modes of Pseudorabies Virus Neuroinvasion and Lethality in Mice. J. Virol. 2004, 78, 12951–12963. [Google Scholar] [CrossRef]
- Taylor, M.P.; Enquist, L.W. Axonal spread of neuroinvasive viral infections. Trends Microbiol. 2015, 23, 283–288. [Google Scholar] [CrossRef]
- Khairallah, M.; Ben Yahia, S.; Ladjimi, A.; Zeghidi, H.; Ben Romdhane, F.; Besbes, L.; Zaouali, S.; Messaoud, R. Chorioretinal involvement in patients with West Nile virus infection. Ophthalmology 2004, 111, 2065–2070. [Google Scholar] [CrossRef]
- Fang, S.-T.; Chu, S.-Y.; Lee, Y.-C. Ischaemic maculopathy in japanese encephalitis. Eye 2006, 20, 1439–1441. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Farr, D.; Kumar, A. Ocular Manifestations of Emerging Flaviviruses and the Blood-Retinal Barrier. Viruses 2018, 10, 530. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, L.; Albecka, A.; Mallery, D.L.; Kellner, M.J.; Paul, D.; Carter, A.P.; James, L.C.; Lancaster, M.A. SARS-CoV-2 Infects the Brain Choroid Plexus and Disrupts the Blood-CSF Barrier in Human Brain Organoids. Cell Stem Cell 2020, 27, 951–961.e5. [Google Scholar] [CrossRef]
- Lyck, R.; Engelhardt, B. Going Against the Tide—How Encephalitogenic T Cells Breach the Blood-Brain Barrier. J. Vasc. Res. 2012, 49, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Jean, C.M.; Honarmand, S.; Louie, J.K.; Glaser, C.A. Risk Factors for West Nile Virus Neuroinvasive Disease, California, 2005. Emerg. Infect. Dis. 2007, 13, 1918–1920. [Google Scholar] [CrossRef]
- Cho, H.; Proll, S.C.; Szretter, K.; Katze, M.G.; Gale, M.; Diamond, M.S. Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nat. Med. 2013, 19, 458–464. [Google Scholar] [CrossRef]
- Liou, M.-L.; Hsu, C.-Y. Japanese encephalitis virus is transported across the cerebral blood vessels by endocytosis in mouse brain. Cell Tissue Res. 1998, 293, 389–394. [Google Scholar] [CrossRef]
- Li, F.; Wang, Y.; Yu, L.; Cao, S.; Wang, K.; Yuan, J.; Wang, C.; Wang, K.; Cui, M.; Fu, Z.F. Viral Infection of the Central Nervous System and Neuroinflammation Precede Blood-Brain Barrier Disruption during Japanese Encephalitis Virus Infection. J. Virol. 2015, 89, 5602–5614. [Google Scholar] [CrossRef]
- Hsieh, J.T.; Rathore, A.P.S.; Soundarajan, G.; John, A.L.S. Japanese encephalitis virus neuropenetrance is driven by mast cell chymase. Nat. Commun. 2019, 10, 706. [Google Scholar] [CrossRef]
- Růžek, D.; Salát, J.; Singh, S.K.; Kopecký, J. Breakdown of the Blood-Brain Barrier during Tick-Borne Encephalitis in Mice Is Not Dependent on CD8+ T-Cells. PLoS ONE 2011, 6, e20472. [Google Scholar] [CrossRef] [Green Version]
- Chekhonin, V.P.; Zhirkov, Y.A.; Belyaeva, I.A.; Ryabukhin, I.A.; Gurina, O.I.; Dmitriyeva, T.B. Serum time course of two brain-specific proteins, α1 brain globulin and neuron-specific enolase, in tick-born encephalitis and Lyme disease. Clin. Chim. Acta 2002, 320, 117–125. [Google Scholar] [CrossRef]
- Vodička, J.; Jelínková, H.; Chrobok, V. Smell impairment after tick-borne encephalitis vaccination: Case report. Vaccine 2010, 28, 886–888. [Google Scholar] [CrossRef] [PubMed]
- Voulgari, N.; Blanc, C.-M.; Guido, V.; Rossi, D.C.; Guex-Crosier, Y.; Hoogewoud, F. Tick-borne encephalitis related uveitis: A case report. BMC Ophthalmol. 2021, 21, 315. [Google Scholar] [CrossRef] [PubMed]
- Benzarti, E.; Sarlet, M.; Franssen, M.; Desmecht, D.; Schmidt-Chanasit, J.; Garigliany, M.-M. New Insights into the Susceptibility of Immunocompetent Mice to Usutu Virus. Viruses 2020, 12, 189. [Google Scholar] [CrossRef] [PubMed]
- Alimonti, J.B.; Ribecco-Lutkiewicz, M.; Sodja, C.; Jezierski, A.; Stanimirovic, D.B.; Liu, Q.; Haqqani, A.S.; Conlan, W.; Bani-Yaghoub, M. Zika virus crosses an in vitro human blood brain barrier model. Fluids Barriers CNS 2018, 15, 15. [Google Scholar] [CrossRef]
- Ayala-Nunez, N.V.; Follain, G.; Delalande, F.; Hirschler, A.; Partiot, E.; Hale, G.L.; Bollweg, B.C.; Roels, J.; Chazal, M.; Bakoa, F.; et al. Zika virus enhances monocyte adhesion and transmigration favoring viral dissemination to neural cells. Nat. Commun. 2019, 10, 4430. [Google Scholar] [CrossRef]
- de Carvalho, G.C.; Borget, M.-Y.; Bernier, S.; Garneau, D.; Duarte, A.J.D.S.; Dumais, N. RAGE and CCR7 mediate the transmigration of Zika-infected monocytes through the blood-brain barrier. Immunobiology 2019, 224, 792–803. [Google Scholar] [CrossRef]
- Ozdener, M.H.; Donadoni, M.; Cicalese, S.; Spielman, A.I.; Garcia-Blanco, A.; Gordon, J.; Sariyer, I.K. Zika virus infection in chemosensory cells. J. Neurovirol. 2020, 26, 371–381. [Google Scholar] [CrossRef]
- Lazarini, F.; Lannuzel, A.; Cabié, A.; Michel, V.; Madec, Y.; Chaumont, H.; Calmont, I.; Favrat, M.; Montagutelli, X.; Roze, E.; et al. Olfactory outcomes in Zika virus-associated Guillain–Barré syndrome. Eur. J. Neurol. 2022, 29, 2823–2831. [Google Scholar] [CrossRef]
- Fernandes, N.C.; Nogueira, J.S.; Réssio, R.A.; Cirqueira, C.S.; Kimura, L.M.; Fernandes, K.R.; Cunha, M.S.; Souza, R.P.; Guerra, J.M. Experimental Zika virus infection induces spinal cord injury and encephalitis in newborn Swiss mice. Exp. Toxicol. Pathol. 2017, 69, 63–71. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Family | Genus | Species |
|---|---|---|
| Flaviviridae | Flavivirus | West Nile virus |
| Usutu virus | ||
| Japanese encephalitis virus | ||
| Saint Louis encephalitis virus | ||
| Murray Valley encephalitis virus | ||
| Ilheus virus | ||
| Zika virus | ||
| Wesselsbron virus | ||
| Dengue virus | ||
| Powassan virus | ||
| Tick-borne encephalitis virus |
| Role | ||||
|---|---|---|---|---|
| Type | Factor | Virus | Increase Invasion | Decrease Invasion |
| Adhesion molecules | E-selectin | WNV [5] | Increases recruitment of leukocytes to the BBB and CNS and enhances attachment of leukocytes. Allows for potential Trojan horse and further release of inflammatory cytokines that impact haematogenous barrier integrity. | |
| ICAM-1 | JEV [6] WNV [5,71] | |||
| VCAM-1 | WNV [5] | |||
| Chemokine | CINC-1 | JEV [6] | ||
| CXCL9 | WNV [69] | |||
| CXCL10 | WNV [69] | |||
| MCP-1 | JEV [46] | |||
| MCP-5 | WNV [69] | |||
| N51/KC | MVEV [70] | |||
| Osteopontin | WNV [75] | |||
| RANTES | JEV [6] | |||
| Cytokine | IFN-γ | WNV [38] | Activates and disrupts the BBB. | |
| IL-1α | JEV [76] | |||
| IL1-β | ZIKV [34] | |||
| TNF-α | WNV [60] | |||
| Enzyme | Cathepsin L | ZIKV JEV WNV DENV [61] | Disrupts EGL layer of brain endothelium [61]. | |
| Endoglycosidase heparinase | ||||
| Sialidases | ||||
| MMP-9 | JEV [40], TBEV [41] WNV [42,43] | Involved in degradation of BBB TJ proteins [42,43]. | ||
| RacA | Down regulates RhoA [37]. | |||
| RhoA | Hyper activation leads to junctional disruption [37]. | |||
| Receptor | CLEC5a | JEV WNV [46] | Blockage preserved BBB integrity [46] indicating role in BBB dysregulation via induction of inflammatory mediator release [46]. | |
| GAGs | WNV [14]. JEV MVEV [16,17] | Attachment receptor [14,15]. | Increased GAG binding sequesters virus in periphery [17,18,19]. | |
| IFNAR | WNV [38] | Balances RhoA-RacA activation [37]. Increases localisation of TJ proteins at endothelial cell border [38]. | ||
| IFNLR1 | WNV [39] | KO increases permeability of BBB indicating role in BBB maintenance [39]. | ||
| MFSD2A | ZIKV [13] | Inhibits vesicular transcytosis across BMECs [77] and is ubiquitinated by ZIKV E protein binding [13]. | ||
| TAM | ZIKV [32] DENV [33] WNV [35] JEV [36] | Candidate entry receptor for ZIKV and DENV [32,33]. | Involved in stabilisation of endothelial TJs [35,36]. | |
| Haematogenous | Transneural | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Virus | Infection of BMECs * | Transport across BMECs * | Trojan Horse | Increased BBB Permeability | Choroid Plexus | Olfactory | Spinal Cord | Gut–Brain | Optical Nerve |
| JEV | ✓ [6,119] | ✓ [119] | X [11,29] | ✓ [31,46,61,120] | X [29] | ✓ [29,97,98] | ? [121] | ? | ? [113] |
| TBEV | ✓ [8] | ✓ [8] | ? | ✓ [41,122,123] | ? | ? [124] | ? [85,86] | ? [103,105] | ? [125] |
| USUV | ✓ [23] | ✓ [23] | ? | X [23] | ? | ? [126] | ? [23] | ? | ? [23] |
| WNV | ✓ [5,12] | ✓ [5,14] | ✓ [71,73] | ✓ [10,42,43,61] | ✓ [28] | ✓ [91,92,93] | ✓ [78,79,81,92] | ? [92,107] | ? [112] |
| ZIKV | ✓ [13] | ✓ [127] | ✓ [128,129] | ✓ [61] | ✓ [27] | ? [130,131] | ? [132] | ? | ? [114] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marshall, E.M.; Koopmans, M.P.G.; Rockx, B. A Journey to the Central Nervous System: Routes of Flaviviral Neuroinvasion in Human Disease. Viruses 2022, 14, 2096. https://doi.org/10.3390/v14102096
Marshall EM, Koopmans MPG, Rockx B. A Journey to the Central Nervous System: Routes of Flaviviral Neuroinvasion in Human Disease. Viruses. 2022; 14(10):2096. https://doi.org/10.3390/v14102096
Chicago/Turabian StyleMarshall, Eleanor M., Marion P. G. Koopmans, and Barry Rockx. 2022. "A Journey to the Central Nervous System: Routes of Flaviviral Neuroinvasion in Human Disease" Viruses 14, no. 10: 2096. https://doi.org/10.3390/v14102096
APA StyleMarshall, E. M., Koopmans, M. P. G., & Rockx, B. (2022). A Journey to the Central Nervous System: Routes of Flaviviral Neuroinvasion in Human Disease. Viruses, 14(10), 2096. https://doi.org/10.3390/v14102096