

First Polycipivirus and Unmapped RNA Virus Diversity in the Yellow Crazy Ant, Anoplolepis gracilipes

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection, RNA Purification, and Sequencing
2.2. De Novo Assembly and Virus Characterization
2.3. Virus Verification and Field Virus Prevalence
2.4. Phylogenetic and Evolutionary Analysis
3. Results
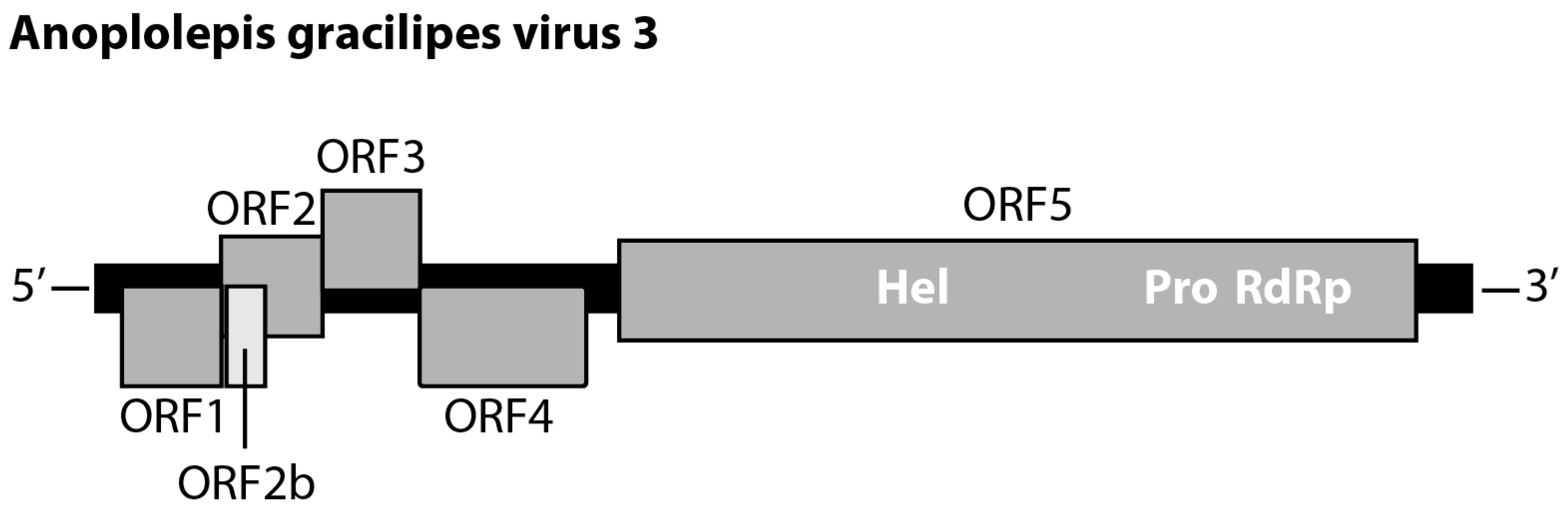
3.1. Characterization of the Virus Genome
3.2. Virus Identification
3.2.1. Polycipivirus
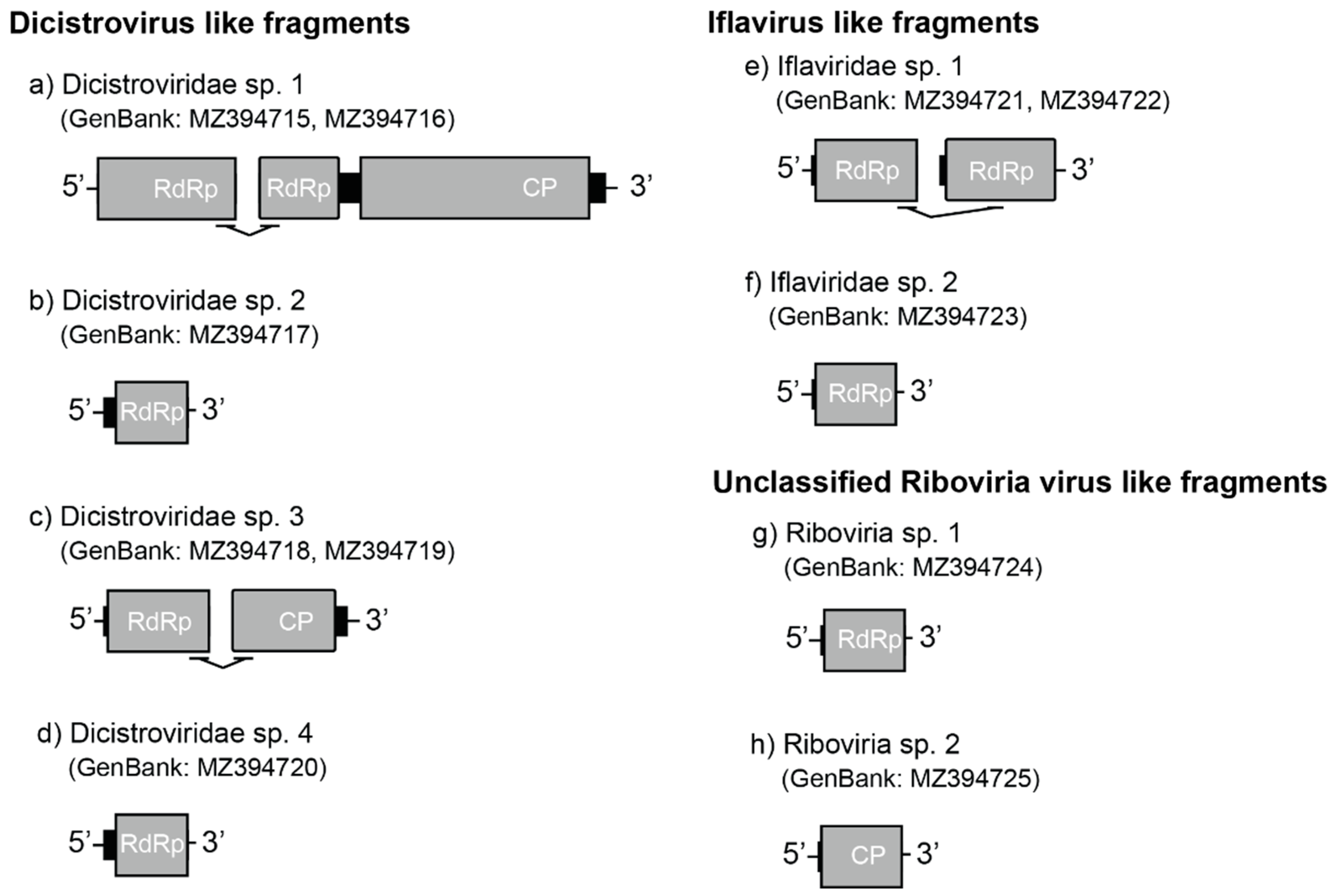
3.2.2. Dicistrovirus-like Fragments
3.2.3. Iflavirus-like Fragments
3.2.4. Unclassified Riboviria-like Fragments
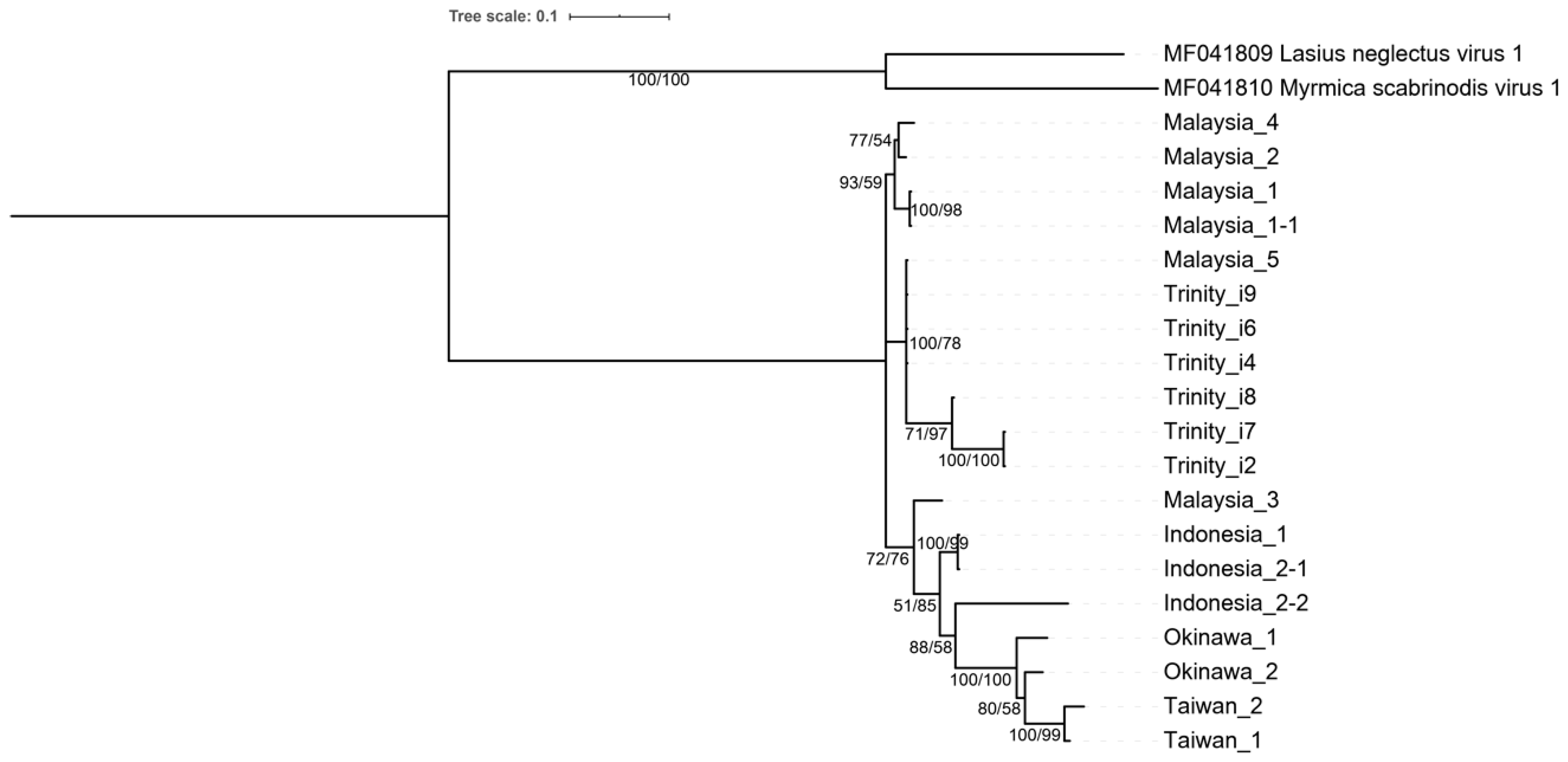
3.3. Phylogenetic Analysis and Polycipivirus Classification
3.4. Field Prevalence and Evolutionary Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Wilgenburg, E.; Torres, C.W.; Tsutsui, N.D. The global expansion of a single ant supercolony. Evol. Appl. 2010, 3, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Eyer, P.A.; McDowell, B.; Johnson, L.N.L.; Calcaterra, L.A.; Fernandez, M.B.; Shoemaker, D.; Puckett, R.T.; Vargo, E.L. Supercolonial structure of invasive populations of the tawny crazy ant Nylanderia fulva in the US. BMC Evol. Biol. 2018, 18, 209. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, B.D.; Hagedorn, H. Quantification of supercolonial traits in the yellow crazy ant, Anoplolepis gracilipes. J. Insect Sci. 2014, 14, 25. [Google Scholar] [CrossRef]
- Cremer, S. Pathogens and disease defense of invasive ants. Curr. Opin. Insect Sci. 2019, 33, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Tragust, S.; Feldhaar, H.; Espadaler, X.; Pedersen, J.S. Rapid increase of the parasitic fungus Laboulbenia formicarum in supercolonies of the invasive garden ant Lasius neglectus. Biol. Invasions 2015, 17, 2795–2801. [Google Scholar] [CrossRef]
- Ugelvig, L.V.; Cremer, S. Effects of social immunity and unicoloniality on host-parasite interactions in invasive insect societies. Funct. Ecol. 2012, 26, 1300–1313. [Google Scholar] [CrossRef]
- Brahma, A.; Leon, R.G.; Hernandez, G.L.; Wurm, Y. Larger, more connected societies of ants have a higher prevalence of viruses. Mol. Ecol. 2022, 31, 859–865. [Google Scholar] [CrossRef]
- Allen, C.; Valles, S.M.; Strong, C.A. Multiple virus infections occur in individual polygyne and monogyne Solenopsis invicta ants. J. Invertebr. Pathol. 2011, 107, 107–111. [Google Scholar] [CrossRef]
- Valles, S.M.; Varone, L.; Ramírez, L.; Briano, J. Multiplex detection of Solenopsis invicta viruses-1, -2, and -3. J. Virol. Methods 2009, 162, 276–279. [Google Scholar] [CrossRef]
- Viljakainen, L.; Holmberg, I.; Abril, S.; Jurvansuu, J. Viruses of invasive Argentine ants from the European main supercolony: Characterization, interactions and evolution. J. Gen. Virol. 2018, 99, 1129–1140. [Google Scholar] [CrossRef]
- Sébastien, A.; Lester, P.J.; Hall, R.J.; Wang, J.; Moore, N.E.; Gruber, M.A. Invasive ants carry novel viruses in their new range and form reservoirs for a honeybee pathogen. Biol. Lett. 2015, 11, 20150610. [Google Scholar] [CrossRef] [PubMed]
- Gruber, M.A.M.; Cooling, M.; Baty, J.W.; Buckley, K.; Friedlander, A.; Quinn, O.; Russell, J.F.E.J.; Sébastien, A.; Lester, P.J. Single-stranded RNA viruses infecting the invasive Argentine ant, Linepithema humile. Sci. Rep. 2017, 7, 3304. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Yang, C.C.S. Biology, ecology, and management of the invasive longlegged ant, Anoplolepis gracilipes. Annu. Rev. Entomol. 2022, 67, 43–63. [Google Scholar] [CrossRef] [PubMed]
- Ito, F.; Asfiya, W.; Kojima, J.I. Discovery of independent-founding solitary queens in the yellow crazy ant Anoplolepis gracilipes in East Java, Indonesia (Hymenoptera: Formicidae). Entomol. Sci. 2016, 19, 312–314. [Google Scholar] [CrossRef]
- Hsu, H.W.; Chiu, M.C.; Lee, C.C.; Lee, C.Y.; Yang, C.C.S. The association between virus prevalence and intercolonial aggression levels in the yellow crazy ant, Anoplolepis gracilipes (Jerdon). Insects 2019, 10, 436. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Lin, C.Y.; Tseng, S.P.; Matsuura, K.; Yang, C.C.S. Ongoing coevolution of Wolbachia and a widespread invasive ant, Anoplolepis gracilipes. Microorganisms 2020, 8, 1569. [Google Scholar] [CrossRef]
- Lester, P.J.; Sébastien, A.; Suarez, A.V.; Barbieri, R.F.; Gruber, M.A. Symbiotic bacterial communities in ants are modified by invasion pathway bottlenecks and alter host behavior. Ecology 2017, 98, 861–874. [Google Scholar] [CrossRef]
- Lee, C.C.; Lin, C.Y.; Hsu, H.W.; Yang, C.C.S. Complete genome sequences of two novel dicistroviruses detected in the yellow crazy ant, Anoplolepis gracilipes. Arch. Virol. 2020, 165, 2715–2719. [Google Scholar] [CrossRef]
- Cooling, M.; Gruber, M.A.M.; Hoffmann, B.D.; Sébastien, A.; Lester, P.J. A metatranscriptomic survey of the invasive yellow crazy ant, Anoplolepis gracilipes, identifies several potential viral and bacterial pathogens and mutualists. Insectes Soc. 2017, 64, 197–207. [Google Scholar] [CrossRef]
- Xavier, C.A.; Allen, M.L.; Whitfield, A.E. Ever-increasing viral diversity associated with the red imported fire ant Solenopsis invicta (Formicidae: Hymenoptera). Virol. J. 2021, 18, 5. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.J.; Tappu, R. MEGAN community edition-interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef]
- Rombel, I.T.; Sykes, K.F.; Rayner, S.; Johnston, S.A. ORF-FINDER: A vector for high-throughput gene identification. Gene 2002, 282, 33–41. [Google Scholar] [CrossRef]
- Söding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Yang, Z. PAMLX: A graphical user interface for PAML. Mol. Biol. Evol. 2013, 30, 2723–2724. [Google Scholar] [CrossRef]
- Sanborn, M.A.; Klein, T.A.; Kim, H.C.; Fung, C.K.; Figueroa, K.L.; Yang, Y.; Asafo-Adjei, E.A.; Jarman, R.G.; Hang, J. Metagenomic analysis reveals three novel and prevalent mosquito viruses from a single pool of Aedes vexans nipponii Collected in the Republic of Korea. Viruses 2019, 11, 222. [Google Scholar] [CrossRef] [Green Version]
- Temmam, S.; Hul, V.; Bigot, T.; Hoem, T.; Gorman, C.; Duong, V.; Dussart, P.; Cappelle, J.; Eloit, M. A novel Polycipiviridae virus identified in Pteropus lylei stools. Microbiol. Resour. Announc. 2019, 8, e01662-18. [Google Scholar] [CrossRef]
- Wright, A.A.; Cross, A.R.; Harper, S.J. A bushel of viruses: Identification of seventeen novel putative viruses by RNA-seq in six apple trees. PLoS ONE 2020, 15, e0227669. [Google Scholar] [CrossRef] [PubMed]
- Baty, J.W.; Bulgarella, M.; Dobelmann, J.; Felden, A.; Lester, P.J. Viruses and their effects in ants (Hymenoptera: Formicidae). Myrmecol. News 2020, 30, 213–228. [Google Scholar]
- Koyama, S.; Urayama, S.I.; Ohmatsu, T.; Sassa, Y.; Sakai, C.; Takata, M.; Hayashi, S.; Nagai, M.; Furuya, T.; Moriyama, H.; et al. Identification, characterization and full-length sequence analysis of a novel dsRNA virus isolated from the arboreal ant Camponotus yamaokai. J. Gen. Virol. 2015, 96, 1930–1937. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Sakai, C.; Thomas, C.E.; Nunoura, T.; Urayama, S.I. A new member of the family Totiviridae associated with arboreal ants (Camponotus nipponicus). Arch. Virol. 2016, 161, 2043–2045. [Google Scholar] [CrossRef]
- Rosario, K.; Mettel, K.A.; Benner, B.E.; Johnson, R.; Scott, C.; Yusseff-Vanegas, S.Z.; Baker, C.C.; Cassill, D.L.; Storer, C.; Varsani, A.; et al. Virus discovery in all three major lineages of terrestrial arthropods highlights the diversity of single-stranded DNA viruses associated with invertebrates. PeerJ 2018, 6, e5761. [Google Scholar] [CrossRef]
- Valles, S.M.; Shoemaker, D.; Wurm, Y.; Strong, C.A.; Varone, L.; Becnel, J.J.; Shirk, P.D. Discovery and molecular characterization of an ambisense densovirus from South American populations of Solenopsis invicta. Biol. Control 2013, 67, 431–439. [Google Scholar] [CrossRef]
- Olendraite, I.; Lukhovitskaya, N.I.; Porter, S.D.; Valles, S.M.; Firth, A.E. Polycipiviridae: A proposed new family of polycistronic picorna-like RNA viruses. J. Gen. Virol. 2017, 98, 2368. [Google Scholar] [CrossRef]
- Olendraite, I.; Brown, K.; Valles, S.M.; Firth, A.E.; Chen, Y.; Guérin, D.M.; Hashimoto, Y.; Herrero, S.; de Miranda, J.R.; Ryabov, E.; et al. ICTV virus taxonomy profile: Polycipiviridae. J. Gen. Virol. 2019, 100, 554. [Google Scholar] [CrossRef]
- Valles, S.M.; Strong, C.A.; Hashimoto, Y. A new positive-strand RNA virus with unique genome characteristics from the red imported fire ant, Solenopsis invicta. Virology 2007, 365, 457–463. [Google Scholar] [CrossRef]
- Valles, S.M.; Rivers, A.R. Nine new RNA viruses associated with the fire ant Solenopsis invicta from its native range. Virus Genes 2019, 55, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, F.; Hirai, M.; Takaki, Y.; Shimane, Y.; Thomas, C.E.; Urayama, S.I.; Nunoura, T.; Koyama, S. A new polycipivirus identified in Colobopsis shohki. Arch. Virol. 2020, 165, 761–763. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Manfredini, F.; Shoemaker, D.; Grozinger, C.M. Dynamic changes in host–virus interactions associated with colony founding and social environment in fire ant queens (Solenopsis invicta). Ecol. Evol. 2016, 6, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S.; Andino, R. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog. 2010, 6, e1001005. [Google Scholar] [CrossRef]
- Wetterer, J.K. Worldwide distribution and potential spread of the longlegged ant, Anoplolepis gracilipes (Hymenoptera: Formicidae). Sociobiology 2005, 45, 77–97. [Google Scholar]
- Chinchio, E.; Crotta, M.; Romeo, C.; Drewe, J.A.; Guitian, J.; Ferrari, N. Invasive alien species and disease risk: An open challenge in public and animal health. PLoS Pathog. 2020, 16, e1008922. [Google Scholar] [CrossRef]
- Collins, L.M.; Warnock, N.D.; Tosh, D.G.; McInnes, C.; Everest, D.; Montgommery, W.I.; Scantlebury, M.; Marks, N.; Dick, J.T.; Reid, N. Squirrelpox virus: Assessing prevalence, transmission and environmental degradation. PLoS ONE 2014, 9, e89521. [Google Scholar] [CrossRef]
- Alger, S.A.; Burnham, P.A.; Boncristiani, H.F.; Brody, A.K. RNA virus spillover from managed honeybees (Apis mellifera) to wild bumblebees (Bombus spp.). PLoS ONE 2019, 14, e0217822. [Google Scholar] [CrossRef]
- Nanetti, A.; Bortolotti, L.; Cilia, G. Pathogens spillover from honey bees to other arthropods. Pathogens 2021, 10, 1044. [Google Scholar] [CrossRef]
- Allen, M.L. Near-Complete Genome Sequences of New Strains of Nylanderia Fulva Virus 1 from Solenopsis invicta. Microbiol. Resour. Announc. 2020, 9, e00798-19. [Google Scholar] [CrossRef] [PubMed]
- Valles, S.M.; Oi, D.H.; Oliver, J.B.; Becnel, J.J. Characterization of Solenopsis invicta virus 4, a polycipivirus infecting the red imported fire ant Solenopsis invicta. Arch. Virol. 2022, in press. [Google Scholar] [CrossRef]
- Yang, C.C.; Yu, Y.C.; Valles, S.M.; Oi, D.H.; Chen, Y.C.; Shoemaker, D.; Wu, W.J.; Shih, C.J. Loss of microbial (pathogen) infections associated with recent invasions of the red imported fire ant Solenopsis invicta. Biol. Invasions 2010, 12, 3307–3318. [Google Scholar] [CrossRef]
- Byrne, A.; Cole, C.; Volden, R.; Vollmers, C. Realizing the potential of full-length transcriptome sequencing. Philos. Trans. R. Soc. B 2019, 374, 20190097. [Google Scholar] [CrossRef] [PubMed]
- Villamor, D.E.V.; Ho, T.; Al Rwahnih, M.; Martin, R.R.; Tzanetakis, I.E. High throughput sequencing for plant virus detection and discovery. Phytopathology 2019, 109, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family/Realm | Genus | Species | Average Read Depth * | Contig Number | NCBI Accessions |
|---|---|---|---|---|---|
| Polycipiviridae | Sopolycivirus | Anoplolepis gracilipes virus 3 | 95.73 | 18 | MW078933 |
| Dicistroviridae | Triatovirus | Anoplolepis gracilipes virus 1 | 50,371.26 | 2 | MT108239 + |
| Dicistroviridae | Triatovirus | Anoplolepis gracilipes virus 2 | 42.63 | 3 | MT108240 + |
| Dicistroviridae | unclassified | Dicistroviridae sp. 1 | 170.87 | 2 | MZ394715 MZ394716 |
| Dicistroviridae | unclassified | Dicistroviridae sp. 2 | 5.77 | 1 | MZ394717 |
| Dicistroviridae | unclassified | Dicistroviridae sp. 3 | 19.98 | 2 | MZ394718 MZ394719 |
| Dicistroviridae | unclassified | Dicistroviridae sp. 4 | 28.53 | 1 | MZ394720 |
| Iflaviridae | unclassified | Iflaviridae sp. 1 | 3.52 | 2 | MZ394721 MZ394722 |
| Iflaviridae | unclassified | Iflaviridae sp. 2 | 23 | 1 | MZ394723 |
| Riboviria | unclassified | Riboviria sp. 1 | 3.92 | 1 | MZ394724 |
| Riboviria | unclassified | Riboviria sp. 2 | 7.3 | 1 | MZ394725 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.-C.; Hsu, H.-W.; Lin, C.-Y.; Gustafson, N.; Matsuura, K.; Lee, C.-Y.; Yang, C.-C.S. First Polycipivirus and Unmapped RNA Virus Diversity in the Yellow Crazy Ant, Anoplolepis gracilipes. Viruses 2022, 14, 2161. https://doi.org/10.3390/v14102161
Lee C-C, Hsu H-W, Lin C-Y, Gustafson N, Matsuura K, Lee C-Y, Yang C-CS. First Polycipivirus and Unmapped RNA Virus Diversity in the Yellow Crazy Ant, Anoplolepis gracilipes. Viruses. 2022; 14(10):2161. https://doi.org/10.3390/v14102161
Chicago/Turabian StyleLee, Chih-Chi, Hung-Wei Hsu, Chun-Yi Lin, Nicolas Gustafson, Kenji Matsuura, Chow-Yang Lee, and Chin-Cheng Scotty Yang. 2022. "First Polycipivirus and Unmapped RNA Virus Diversity in the Yellow Crazy Ant, Anoplolepis gracilipes" Viruses 14, no. 10: 2161. https://doi.org/10.3390/v14102161