
HIV-1 Tat Upregulates the Receptor for Advanced Glycation End Products and Superoxide Dismutase-2 in the Heart of Transgenic Mice
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Intracellular Calcium Imaging
2.2. Animal and Housing
2.3. HIV-1 Tat Induction
2.4. Echocardiography
2.5. Tissue Collection
2.5.1. Cardiac Histology
2.5.2. Western Blot
2.5.3. RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction
2.6. Statistical Analyses
3. Results
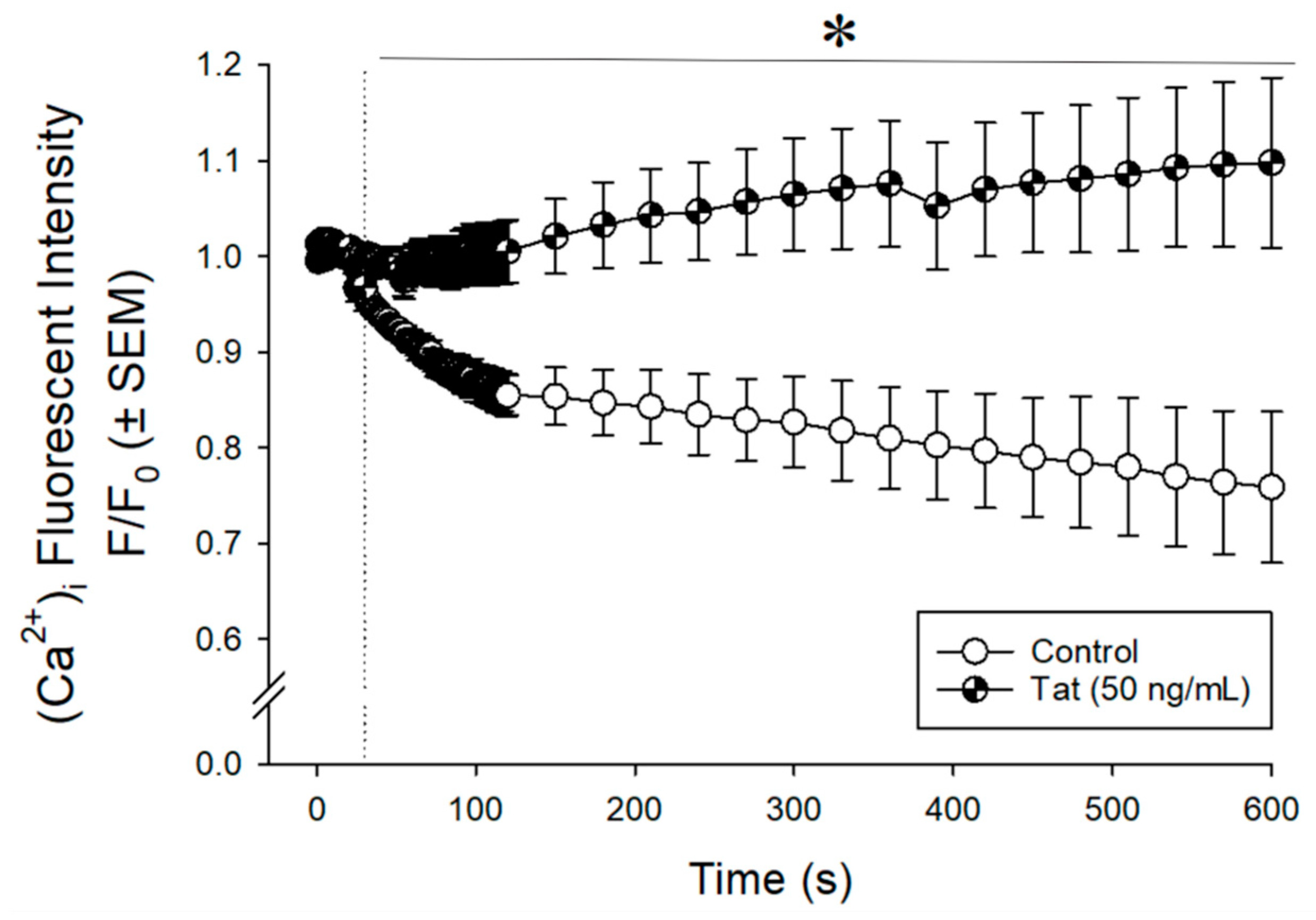
3.1. Tat Dysregulated Intracellular Calcium ([Ca2+]i) in Cultured Human Ventricular Cells
3.2. Tat Induction Increased Tat mRNA Levels in the Whole Heart among Adult Male Mice
3.3. Tat Induction Did Not Alter Cardiac Function among Adult Mice
3.4. Tat Induction Upregulated RAGE and SOD-2 in Ventricular Tissue of Adult Mice
3.5. Tat Exposure Altered Mast Cell Numbers and Collagen Deposition within Ventricular Tissue of Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saylor, D.; Dickens, A.; Sacktor, N.; Haughey, N.; Slusher, B.; Pletnikov, M.; Mankowski, J.L.; Brown, A.; Volsky, D.J.; McArthur, J.C. HIV-associated neurocognitive disorder—Pathogenesis and prospects for treatment. Nat. Rev. Neurol. 2016, 12, 234–248. [Google Scholar] [CrossRef]
- Thakur, K.T.; Boubour, A.; Saylor, D.; Das, M.; Bearden, D.R.; Birbeck, G.L. Global HIV neurology. AIDS 2019, 33, 163–184. [Google Scholar] [CrossRef] [PubMed]
- Veenhuis, R.T.; Clements, J.E.; Gama, L. HIV Eradication Strategies: Implications for the Central Nervous System. Curr. HIV/AIDS Rep. 2019, 16, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Guaraldi, G.; Orlando, G.; Zona, S.; Menozzi, M.; Carli, F.; Garlassi, E.; Berti, A.; Rossi, E.; Roverato, A.; Palella, F. Premature Age-Related Comorbidities Among HIV-Infected Persons Compared with the General Population. Clin. Infect. Dis. 2011, 53, 1120–1126. [Google Scholar] [CrossRef] [Green Version]
- Onen, N.F.; Overton, E.T. A review of premature frailty in HIV-infected persons; another manifestation of HIV-related accelerated aging. Curr. Aging Sci. 2011, 1, 33–41. [Google Scholar] [CrossRef]
- Pathai, S.; Bajillan, H.; Landay, A.L.; High, K.P. Is HIV a Model of Accelerated or Accentuated Aging? J. Gerontol. Ser. A 2014, 69, 833–842. [Google Scholar] [CrossRef] [Green Version]
- Sackoff, J.E.; Hanna, D.B.; Pfeiffer, M.R.; Torian, L.V. Causes of death among persons with AIDS in the era of highly active antiretroviral therapy: New York City. Ann. Intern. Med. 2006, 6, 397–406. [Google Scholar] [CrossRef]
- Triant, V.A.; Brown, T.T.; Lee, H.; Grinspoon, S.K. Fracture Prevalence among Human Immunodeficiency Virus (HIV)-InfectedVersusNon-HIV-Infected Patients in a Large U.S. Healthcare System. J. Clin. Endocrinol. Metab. 2008, 93, 3499–3504. [Google Scholar] [CrossRef] [Green Version]
- Tripathy, S.K.; Agrawala, R.K.; Baliarsinha, A. Endocrine alterations in HIV-infected patients. Indian J. Endocrinol. Metab. 2015, 19, 143–147. [Google Scholar] [CrossRef]
- Neuhaus, J.; Jacobs, D.R., Jr.; Baker, J.V.; Calmy, A.; Duprez, D.; La Rosa, A.; Kuller, L.H.; Pett, S.L.; Ristola, M.; Ross, M.J.; et al. Markers of Inflammation, Coagulation, and Renal Function Are Elevated in Adults with HIV Infection. J. Infect. Dis. 2010, 201, 1788–1795. [Google Scholar] [CrossRef]
- Freiberg, M.S.; Chang, C.-C.H.; Skanderson, M.; Patterson, O.V.; DuVall, S.L.; Brandt, C.A.; So-Armah, K.A.; Vasan, R.S.; Oursler, K.A.; Gottdiener, J.; et al. Association Between HIV Infection and the Risk of Heart Failure with Reduced Ejection Fraction and Preserved Ejection Fraction in the Antiretroviral Therapy Era. JAMA Cardiol. 2017, 2, 536–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monsuez, J.-J.; Escaut, L.; Teicher, E.; Charniot, J.-C.; Vittecoq, D. Cytokines in HIV-associated cardiomyopathy. Int. J. Cardiol. 2007, 120, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Durand, M.; Sheehy, O.; Baril, J.-G.; Lelorier, J.; Tremblay, C.L. Association Between HIV Infection, Antiretroviral Therapy, and Risk of Acute Myocardial Infarction: A Cohort and Nested Case–Control Study Using Québec’s Public Health Insurance Database. JAIDS J. Acquir. Immune Defic. Syndr. 2011, 57, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.; Mary-Krause, M.; Cotte, L.; Gilquin, J.; Partisani, M.; Simon, A.; Boccara, F.; Costagliola, D. Impact of Individual Antiretroviral Drugs on the Risk of Myocardial Infarction in Human Immunodeficiency Virus–Infected PatientsA Case-Control Study Nested Within the French Hospital Database on HIV ANRS Cohort CO4Antiretroviral Drugs, Risk of MI, and HIV. Arch. Intern. Med. 2010, 170, 1228–1238. [Google Scholar] [CrossRef]
- Kaplan, R.C.; Kingsley, L.A.; Sharrett, A.R.; Li, X.; Lazar, J.; Tien, P.C.; Mack, W.J.; Cohen, M.H.; Jacobson, L.; Gange, S.J. Ten-Year Predicted Coronary Heart Disease Risk in HIV-Infected Men and Women. Clin. Infect. Dis. 2007, 45, 1074–1081. [Google Scholar] [CrossRef] [Green Version]
- Savès, M.; Chêne, G.; Ducimetière, P.; Leport, C.; Le Moal, G.; Amouyel, P.; Arveiler, D.; Ruidavets, J.-B.; Reynes, J.; Bingham, A.; et al. Risk Factors for Coronary Heart Disease in Patients Treated for Human Immunodeficiency Virus Infection Compared with the General Population. Clin. Infect. Dis. 2003, 37, 292–298. [Google Scholar] [CrossRef] [Green Version]
- Vittecoq, D.; Escaut, L.; Chironi, G.; Teicher, E.; Monsuez, J.J.; Andrejak, M.; Simon, A. Coronary heart disease in HIV-infected patients in the highly active antiretroviral treatment era. AIDS 2003, 17, S70–S76. [Google Scholar] [CrossRef] [Green Version]
- Tseng, Z.H.; Secemsky, E.A.; Dowdy, D.; Vittinghoff, E.; Moyers, B.; Wong, J.K.; Havlir, D.V.; Hsue, P.Y. Sudden Cardiac Death in Patients with Human Immunodeficiency Virus Infection. J. Am. Coll. Cardiol. 2012, 59, 1891–1896. [Google Scholar] [CrossRef] [Green Version]
- Narla, V.A. Sudden cardiac death in HIV-infected patients: A contemporary review. Clin. Cardiol. 2021, 44, 316–321. [Google Scholar] [CrossRef]
- Mensah, G.A.; Roth, G.A.; Fuster, V. The Global Burden of Cardiovascular Diseases and Risk Factors: 2020 and beyond. J. Am. Coll. Cardiol. 2019, 74, 2529–2532. [Google Scholar] [CrossRef]
- Feinstein, M.J.; Hsue, P.Y.; Benjamin, L.; Bloomfield, G.S.; Currier, J.S.; Freiberg, M.S.; Grinspoon, S.K.; Levin, J.; Longenecker, C.T.; Post, W.S. Characteristics, Prevention, and Management of Cardiovascular Disease in People Living With HIV: A Scientific Statement from the American Heart Association. Circulation 2019, 140, e98–e124. [Google Scholar] [CrossRef] [PubMed]
- Smit, M.; Brinkman, K.; Geerlings, S.; Smit, C.; Thyagarajan, K.; van Sighem, A.; de Wolf, F.; Hallett, T.B. Future challenges for clinical care of an ageing population infected with HIV: A modelling study. Lancet Infect. Dis. 2015, 15, 810–818. [Google Scholar] [CrossRef] [Green Version]
- Esser, S.; Gelbrich, G.; Brockmeyer, N.; Goehler, A.; Schadendorf, D.; Erbel, R.; Neumann, T.; Reinsch, N. Prevalence of cardiovascular diseases in HIV-infected outpatients: Results from a prospective, multicenter cohort study. Clin. Res. Cardiol. 2013, 102, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.P.; Patel, K.; Johnson, K.R.; Maric, D.; Calabresi, P.A.; Hasbun, R.; Nath, A. Induction of IL-17 and nonclassical T-cell activation by HIV-Tat protein. Proc. Natl. Acad. Sci. USA 2013, 110, 13588–13593. [Google Scholar] [CrossRef] [Green Version]
- Henderson, L.J.; Johnson, T.P.; Smith, B.R.; Reoma, L.; Santamaria, U.A.; Bachani, M.; DeMarino, C.; Barclay, R.A.; Snow, J.; Sacktor, N.; et al. Presence of Tat and transactivation response element in spinal fluid despite antiretroviral therapy. AIDS 2019, 33 (Suppl. S2), S145–S157. [Google Scholar] [CrossRef]
- Das, A.T.; Harwig, A.; Berkhout, B. The HIV-1 Tat Protein Has a Versatile Role in Activating Viral Transcription. J. Virol. 2011, 85, 9506–9516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Re, M.C.; Furlini, G.; Vignoli, M.; Ramazzotti, E.; Roderigo, G.; De Rosa, V.; Zauli, G.; Lolli, S.; Capitani, S.; La Placa, M. Effect of Antibody to HIV-1 Tat Protein on Viral Replication in Vitro and Progression of HIV-1 Disease in Vivo. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1995, 10, 408–416. [Google Scholar] [CrossRef]
- Liu, K.; Chi, D.S.; Li, C.; Hall, H.K.; Milhorn, D.M.; Krishnaswamy, G. HIV-1 Tat protein-induced VCAM-1 expression in human pulmonary artery endothelial cells and its signaling. Am. J. Physiol. Cell. Mol. Physiol. 2005, 289, L252–L260. [Google Scholar] [CrossRef] [Green Version]
- Duan, M.; Yao, H.; Hu, G.; Chen, X.; Lund, A.K.; Buch, S. HIV Tat Induces Expression of ICAM-1 in HUVECs: Implications for miR-221/-222 in HIV-Associated Cardiomyopathy. PLoS ONE 2013, 8, e60170. [Google Scholar] [CrossRef]
- Zhan, J.; Qin, S.; Lu, L.; Hu, X.; Zhou, J.; Sun, Y.; Yang, J.; Liu, Y.; Wang, Z.; Tan, N.; et al. miR-34a is a common link in both HIV- and antiretroviral therapy-induced vascular aging. Aging 2016, 8, 3298–3310. [Google Scholar] [CrossRef]
- Ben Haij, N.; Planès, R.; Leghmari, K.; Serrero, M.; Delobel, P.; Izopet, J.; BenMohamed, L.; Bahraoui, E. HIV-1 Tat Protein Induces Production of Proinflammatory Cytokines by Human Dendritic Cells and Monocytes/Macrophages through Engagement of TLR4-MD2-CD14 Complex and Activation of NF-κB Pathway. PLoS ONE 2015, 10, e0129425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cota-Gomez, A.; Flores, N.C.; Cruz, C.; Casullo, A.; Aw, T.Y.; Ichikawa, H.; Schaack, J.; Scheinman, R.; Flores, S.C. The Human Immunodeficiency Virus-1 Tat Protein Activates Human Umbilical Vein Endothelial Cell E-selectin Expression via an NF-κB-dependent Mechanism. J. Biol. Chem. 2002, 277, 14390–14399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Z.; Hernandez, R.; Liu, J.; Gwag, T.; Lu, W.; Hsiai, T.K.; Kaul, M.; Zhou, T.; Zhou, C. HIV Protein Tat Induces Macrophage Dysfunction and Atherosclerosis Development in Low-Density Lipoprotein Receptor–Deficient Mice. Cardiovasc. Drugs Ther. 2022, 36, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Tahrir, F.G.; Shanmughapriya, S.; Ahooyi, T.M.; Knezevic, T.; Gupta, M.K.; Kontos, C.D.; McClung, J.M.; Madesh, M.; Gordon, J.; Feldman, A.M.; et al. Dysregulation of mitochondrial bioenergetics and quality control by HIV-1 Tat in cardiomyocytes. J. Cell. Physiol. 2018, 233, 748–758. [Google Scholar] [CrossRef] [PubMed]
- Brailoiu, E.; Deliu, E.; Sporici, R.A.; Benamar, K.; Brailoiu, G.C. HIV-1-Tat excites cardiac parasympathetic neurons of nucleus ambiguus and triggers prolonged bradycardia in conscious rats. Am. J. Physiol. Integr. Comp. Physiol. 2014, 306, R814–R822. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Huang, W.; Jiang, W.; Wu, X.; Ye, B.; Zhou, X. HIV-1 Tat Regulates Occludin and AβTransfer Receptor Expression in Brain Endothelial Cells via Rho/ROCK Signaling Pathway. Oxidative Med. Cell. Longev. 2016, 2016, 4196572. [Google Scholar] [CrossRef] [Green Version]
- Zou, M.; Huang, W.; Jiang, W.; Wu, Y.; Chen, Q. Role of Cav-1 in HIV-1 Tat-Induced Dysfunction of Tight Junctions and Aβ-Transferring Proteins. Oxidative Med. Cell. Longev. 2019, 2019, 3403206. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Wu, Y.; Yu, Y.; Wei, J.; Huang, W. Rho-kinase inhibitor hydroxyfasudil protects against HIV-1 Tat-induced dysfunction of tight junction and neprilysin/Aβ transfer receptor expression in mouse brain microvessels. Mol. Cell. Biochem. 2021, 476, 2159–2170. [Google Scholar] [CrossRef]
- Koyama, Y.; Takeishi, Y.; Niizeki, T.; Suzuki, S.; Kitahara, T.; Sasaki, T.; Kubota, I. Soluble Receptor for Advanced Glycation End Products (RAGE) is a Prognostic Factor for Heart Failure. J. Card. Fail. 2008, 14, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Ramasamy, R.; Schmidt, A.M. Receptor for Advanced Glycation End Products (RAGE) and Implications for the Pathophysiology of Heart Failure. Curr. Heart Fail. Rep. 2012, 9, 107–116. [Google Scholar] [CrossRef]
- Hartog, J.W.L.; Voors, A.A.; Bakker, S.J.L.; Smit, A.J.; Van Veldhuisen, D.J. Advanced glycation end-products (AGEs) and heart failure: Pathophysiology and clinical implications. Eur. J. Heart Fail. 2007, 9, 1146–1155. [Google Scholar] [CrossRef] [PubMed]
- Senatus, L.M.; Schmidt, A.M. The AGE-RAGE Axis: Implications for Age-Associated Arterial Diseases. Front. Genet. 2017, 8, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Wang, K.; Penn, M.S.; Marso, S.P.; Lauer, M.A.; Forudi, F.; Zhou, X.; Qu, W.; Lu, Y.; Stern, D.M.; et al. Receptor for AGE (RAGE) Mediates Neointimal Formation in Response to Arterial Injury. Circulation 2003, 107, 2238–2243. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Zhang, H.; Schmidt, A.M.; Zhang, C. AGE/RAGE produces endothelial dysfunction in coronary arterioles in Type 2 diabetic mice. Am. J. Physiol. Circ. Physiol. 2008, 295, H491–H498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wautier, M.-P.; Chappey, O.; Corda, S.; Stern, D.M.; Schmidt, A.M.; Wautier, J.-L. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E685–E694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burr, S.D.; Harmon, M.B.; Stewart, J.A., Jr. The Impact of Diabetic Conditions and AGE/RAGE Signaling on Cardiac Fibroblast Migration. Front. Cell Dev. Biol. 2020, 8, 112. [Google Scholar] [CrossRef]
- El Kamari, V.; Thomas, A.; Shan, L.; Sattar, A.; Monnier, V.; Howell, S.K.; Beisswenger, P.J.; McComsey, G.A. Advanced Glycation End Products Are Associated with Inflammation and Endothelial Dysfunction in HIV. JAIDS J. Acquir. Immune Defic. Syndr. 2019, 81, e55–e62. [Google Scholar] [CrossRef]
- El Kamari, V.; Rodriguez, K.; Moser, C.; Currier, J.S.; Kelesidis, T.; Stein, J.H.; Brown, T.T.; Howell, S.K.; Beisswenger, P.J.; AMccomsey, G. Advanced Glycation End Products Associated with Cardiometabolic Biomarkers In Treated HIV Infection. Open Forum Infect. Dis. 2021, 8, ofab423. [Google Scholar] [CrossRef]
- Jeong, S.J.; Kim, C.O.; Song, Y.G.; Baek, J.-H.; Kim, S.B.; Jin, S.J.; Ku, N.S.; Han, S.H.; Choi, J.Y.; Lee, H.C.; et al. Low plasma levels of the soluble receptor for advanced glycation end products in HIV-infected patients with subclinical carotid atherosclerosis receiving combined antiretroviral therapy. Atherosclerosis 2011, 219, 778–783. [Google Scholar] [CrossRef]
- Paris, J.J.; Zou, S.; Hahn, Y.K.; Knapp, P.E.; Hauser, K.F. 5α-reduced progestogens ameliorate mood-related behavioral pathology, neurotoxicity, and microgliosis associated with exposure to HIV-1 Tat. Brain Behav. Immun. 2016, 55, 202–214. [Google Scholar] [CrossRef]
- Bruce-Keller, A.J.; Turchan-Cholewo, J.; Smart, E.J.; Geurin, T.; Chauhan, A.; Reid, R.; Xu, R.; Nath, A.; Knapp, P.E.; Hauser, K.F. Morphine causes rapid increases in glial activation and neuronal injury in the striatum of inducible HIV-1 tat transgenic mice. Glia 2008, 56, 1414–1427. [Google Scholar] [CrossRef] [Green Version]
- Eng, L.F.; Ghirnikar, R.S.; Lee, Y.L. Glial Fibrillary Acidic Protein: GFAP-Thirty-One Years (1969–2000). Neurochem. Res. 2000, 25, 1439–1451. [Google Scholar] [CrossRef]
- Kashon, M.L.; Ross, G.W.; O’Callaghan, J.P.; Miller, D.B.; Petrovitch, H.; Burchfiel, C.M.; Sharp, D.S.; Markesbery, W.R.; Davis, D.G.; Hardman, J.; et al. Associations of cortical astrogliosis with cognitive performance and dementia status. J. Alzheimers Dis. 2004, 6, 595–604; discussion 673–681. [Google Scholar] [CrossRef]
- Kikel-Coury, N.L.; Brandt, J.P.; Correia, I.A.; O’Dea, M.R.; DeSantis, D.F.; Sterling, F.; Vaughan, K.; Ozcebe, G.; Zorlutuna, P.; Smith, C.J. Identification of astroglia-like cardiac nexus glia that are critical regulators of cardiac development and function. PLoS Biol. 2021, 19, e3001444. [Google Scholar] [CrossRef]
- Kawano, T.; Oki, T.; Uchida, T.; Iuchi, A.; Ogawa, S.; Hayashi, M.; Fukuda, N.; Mori, H.; Ii, K.; Hizawa, K. Innervation of the mitral valve in normal and prolapsed mitral valves. J. Cardiol. Suppl. 1989, 21, 43–54; discussion 55–56. (In Japanese) [Google Scholar] [PubMed]
- Baldarelli, R.M.; Smith, C.M.; Finger, J.H.; Hayamizu, T.F.; McCright, I.J.; Xu, J.; Shaw, D.R.; Beal, J.S.; Blodgett, O.; Campbell, J.; et al. The mouse Gene Expression Database (GXD): 2021 update. Nucleic Acids Res. 2021, 49, D924–D931. [Google Scholar] [CrossRef]
- Lucchetti, J.; Fracasso, C.; Balducci, C.; Passoni, A.; Forloni, G.; Salmona, M.; Gobbi, M. Plasma and Brain Concentrations of Doxycycline after Single and Repeated Doses in Wild-Type and APP23 Mice. J. Pharmacol. Exp. Ther. 2019, 368, 32–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, W.; Grupp, I.L.; Grupp, G.; Hoit, B.; Morris, R.; Samarel, A.M.; Bruggeman, L.; Klotman, P. Cardiac dysfunction occurs in the HIV-1 transgenic mouse treated with zidovudine. Lab. Investig. 2000, 80, 187–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoit, B.D.; Khoury, S.F.; Kranias, E.G.; Ball, N.; Walsh, R.A. In Vivo Echocardiographic Detection of Enhanced Left Ventricular Function in Gene-Targeted Mice with Phospholamban Deficiency. Circ. Res. 1995, 77, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.A.; Wei, C.-C.; Brower, G.L.; Rynders, P.E.; Hankes, G.H.; Dillon, A.R.; Lucchesi, P.A.; Janicki, J.S.; Dell’Italia, L.J. Cardiac mast cell- and chymase-mediated matrix metalloproteinase activity and left ventricular remodeling in mitral regurgitation in the dog. J. Mol. Cell. Cardiol. 2003, 35, 311–319. [Google Scholar] [CrossRef]
- Erqou, S.; Lodebo, B.T.; Masri, A.; Altibi, A.M.; Echouffo-Tcheugui, J.B.; Dzudie, A.; Ataklte, F.; Choudhary, G.; Bloomfield, G.S.; Wu, W.-C.; et al. Cardiac Dysfunction Among People Living With HIV. JACC Heart Fail. 2019, 7, 98–108. [Google Scholar] [CrossRef]
- Cerrato, E.; D’Ascenzo, F.; Biondi-Zoccai, G.; Calcagno, A.; Frea, S.; Marra, W.G.; Castagno, D.; Omedè, P.; Quadri, G.; Sciuto, F.; et al. Cardiac dysfunction in pauci symptomatic human immunodeficiency virus patients: A meta-analysis in the highly active antiretroviral therapy era. Eur. Heart J. 2013, 34, 1432–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friis-Møller, N.; Weber, R.; Reiss, P.; Thiébaut, R.; Kirk, O.; d’Arminio Monforte, A.; Pradier, C.; Morfeldt, L.; Mateu, S.; Law, M.; et al. Cardiovascular disease risk factors in HIV patients--association with antiretroviral therapy. Results from the DAD study. AIDS 2003, 1179–1193. [Google Scholar] [CrossRef]
- Duprez, D.A.; Neuhaus, J.J.; Kuller, L.L.; Tracy, R.R.; Belloso, W.W.; De Wit, S.; Drummond, F.F.; Lane, H.C.H.; Ledergerber, B.; Lundgren, J.; et al. Inflammation, Coagulation and Cardiovascular Disease in HIV-Infected Individuals. PLoS ONE 2012, 7, e44454. [Google Scholar] [CrossRef]
- Kuller, L.L.; Tracy, R.R.; Belloso, W.W.; De Wit, S.; Drummond, F.F.; Lane, C.H.; Ledergerber, B.; Lundgren, J.; Neuhaus, J.J.; Nixon, D.D.; et al. Inflammatory and Coagulation Biomarkers and Mortality in Patients with HIV Infection. PLoS Med. 2008, 5, e203. [Google Scholar] [CrossRef]
- Green, L.A.; Kim, C.; Gupta, S.K.; Rajashekhar, G.; Rehman, J.; Clauss, M. Pentoxifylline reduces tumor necrosis factor-α and HIV-induced vascular endothelial activation. AIDS Res. Hum. Retrovir. 2012, 10, 1207–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsue, P.Y.; Waters, D.D. Time to Recognize HIV Infection as a Major Cardiovascular Risk Factor. Circulation 2018, 138, 1113–1115. [Google Scholar] [CrossRef] [Green Version]
- Neumann, T.; Woiwod, T.; Neumann, A.; Miller, M.; Von Birgelen, C.; Volbracht, L.; Esser, S.; Brockmeyer, N.; Gerken, G.; Erbel, R. Cardiovascular risk factors and probability for cardiovascular events in HIV-infected patients-part III: Age differences. Eur. J. Med. Res. 2004, 9, 267–272. [Google Scholar]
- Lai, L.; Qiu, H. The Physiological and Pathological Roles of Mitochondrial Calcium Uptake in Heart. Int. J. Mol. Sci. 2020, 21, 7689. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, Å.B.; Gottlieb, R.A. Heart mitochondria: Gates of life and death. Cardiovasc. Res. 2008, 77, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, M.T.; Thorburn, D.R.; Penfold, S.A.; Laskowski, A.; Harcourt, B.E.; Sourris, K.C.; Tan, A.L.; Fukami, K.; Thallas-Bonke, V.; Nawroth, P.P.; et al. RAGE-Induced Cytosolic ROS Promote Mitochondrial Superoxide Generation in Diabetes. J. Am. Soc. Nephrol. 2009, 20, 742–752. [Google Scholar] [CrossRef] [PubMed]
- van Deel, E.D.; Lu, Z.; Xu, X.; Zhu, G.; Hu, X.; Oury, T.D.; Bache, R.J.; Duncker, D.J.; Chen, Y. Extracellular superoxide dismutase protects the heart against oxidative stress and hypertrophy after myocardial infarction. Free Radic. Biol. Med. 2008, 44, 1305–1313. [Google Scholar] [CrossRef] [Green Version]
- Juul, K.; Tybjærg-Hansen, A.; Marklund, S.; Heegaard, N.H.; Steffensen, R.; Sillesen, H.; Jensen, G.; Nordestgaard, B.G. Genetically Reduced Antioxidative Protection and Increased Ischemic Heart Disease Risk. Circulation 2004, 109, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondy, K.E.; Gottdiener, J.; Overton, E.T.; Henry, K.; Bush, T.; Conley, L.; Hammer, J.; Carpenter, C.C.; Kojic, E.; Patel, P.; et al. High Prevalence of Echocardiographic Abnormalities among HIV-infected Persons in the Era of Highly Active Antiretroviral Therapy. Clin. Infect. Dis. 2011, 52, 378–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsue, P.Y.; Hunt, P.W.; Ho, J.E.; Farah, H.H.; Schnell, A.; Hoh, R.; Martin, J.N.; Deeks, S.G.; Bolger, A.F. Impact of HIV Infection on Diastolic Function and Left Ventricular Mass. Circ. Heart Fail. 2010, 3, 132–139. [Google Scholar] [CrossRef] [Green Version]
- Schuster, I.; Thöni, G.J.; Ederhy, S.; Walther, G.; Nottin, S.; Vinet, A.; Boccara, F.; Khireddine, M.; Girard, P.-M.; Mauboussin, J.-M.; et al. Subclinical Cardiac Abnormalities in Human Immunodeficiency Virus–Infected Men Receiving Antiretroviral Therapy. Am. J. Cardiol. 2008, 101, 1213–1217. [Google Scholar] [CrossRef] [PubMed]
- Redfield, M.M.; Jacobsen, S.J.; Burnett, J.C.; Mahoney, D.W.; Bailey, K.R.; Rodeheffer, R.J. Burden of Systolic and Diastolic Ventricular Dysfunction in the Community. JAMA 2003, 289, 194–202. [Google Scholar] [CrossRef]
- Mansoor, A.; Golub, E.T.; Dehovitz, J.; Anastos, K.; Kaplan, R.C.; Lazar, J.M. The Association of HIV Infection with Left Ventricular Mass/Hypertrophy. AIDS Res. Hum. Retrovir. 2009, 25, 475–481. [Google Scholar] [CrossRef]
- McIntosh, R.C. A meta-analysis of HIV and heart rate variability in the era of antiretroviral therapy. Clin. Auton. Res. 2016, 26, 287–294. [Google Scholar] [CrossRef]
- Višković, K.; Židovec Lepej, S.; Gorenec, A.; Grgić, I.; Lukas, D.; Zekan, Š.; Dragobratović, A.; Trupković, M.; Begovac, J. Cardiovascular markers of inflammation and serum lipid levels in HIV-infected patients with undetectable viremia. Sci. Rep. 2018, 8, 6113–19548. [Google Scholar] [CrossRef] [Green Version]
- Nou, E.; Lo, J.; Grinspoon, S.K. Inflammation, immune activation, and cardiovascular disease in HIV. AIDS 2016, 30, 1495–1509. [Google Scholar] [CrossRef] [PubMed]
- Mooney, S.; Tracy, R.; Osler, T.; Grace, C. Elevated Biomarkers of Inflammation and Coagulation in Patients with HIV Are Associated with Higher Framingham and VACS Risk Index Scores. PLoS ONE 2015, 10, e0144312. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.; Kan, H.; Lewis, W.; Chen, F.; Sharma, P.; Finkel, M.S. Dilated Cardiomyopathy in Transgenic Mice Expressing HIV Tat. Cardiovasc. Toxicol. 2009, 9, 39–45. [Google Scholar] [CrossRef] [PubMed]
- McDonough, K.H.; Doumen, C.; Giaimo, M.; Prakash, O. Effects of the HIV-1 Protein Tat on Myocardial Function and Response to Endotoxin. Cardiovasc. Toxicol. 2010, 10, 250–258. [Google Scholar] [CrossRef]
- Raidel, S.M.; Haase, C.; Jansen, N.R.; Russ, R.B.; Sutliff, R.L.; Velsor, L.W.; Day, B.J.; Hoit, B.D.; Samarel, A.M.; Lewis, W. Targeted myocardial transgenic expression of HIV Tat causes cardiomyopathy and mitochondrial damage. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1672–H1678. [Google Scholar] [CrossRef] [Green Version]
- Tahrir, F.G.; Knezevic, T.; Gupta, M.K.; Gordon, J.; Cheung, J.Y.; Feldman, A.M.; Khalili, K. Evidence for the Role of BAG3 in Mitochondrial Quality Control in Cardiomyocytes. J. Cell. Physiol. 2016, 232, 797–805. [Google Scholar] [CrossRef] [Green Version]
- Parihar, P.; Parihar, M.S. Metabolic enzymes dysregulation in heart failure: The prospective therapy. Heart Fail. Rev. 2016, 22, 109–121. [Google Scholar] [CrossRef]
- Fitting, S.; Knapp, P.E.; Zou, S.; Marks, W.; Bowers, M.S.; Akbarali, H.; Hauser, K.F. Interactive HIV-1 Tat and Morphine-Induced Synaptodendritic Injury Is Triggered through Focal Disruptions in Na+ Influx, Mitochondrial Instability, and Ca2+ Overload. J. Neurosci. 2014, 34, 12850–12864. [Google Scholar] [CrossRef] [Green Version]
- Paris, J.J.; Liere, P.; Kim, S.; Mahdi, F.; Buchanan, M.E.; Nass, S.R.; Qrareya, A.N.; Salahuddin, M.F.; Pianos, A.; Fernandez, N.; et al. Pregnane steroidogenesis is altered by HIV-1 Tat and morphine: Physiological allopregnanolone is protective against neurotoxic and psychomotor effects. Neurobiol. Stress 2020, 12, 100211. [Google Scholar] [CrossRef]
- Splawski, I.; Timothy, K.W.; Decher, N.; Kumar, P.; Sachse, F.B.; Beggs, A.H.; Sanguinetti, M.C.; Keating, M.T. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc. Natl. Acad. Sci. USA 2005, 102, 8089–8096; discussion 8086–8088. [Google Scholar] [CrossRef] [Green Version]
- Koczor, C.A.; Fields, E.; Jedrzejczak, M.J.; Jiao, Z.; Ludaway, T.; Russ, R.; Shang, J.; Torres, R.A.; Lewis, W. Methamphetamine and HIV-Tat alter murine cardiac DNA methylation and gene expression. Toxicol. Appl. Pharmacol. 2015, 288, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Liu, R.-M.; Kundu, R.K.; Sangiorgi, F.; Wu, W.; Maxson, R.; Forman, H.J. Molecular Mechanism of Decreased Glutathione Content in Human Immunodeficiency Virus Type 1 Tat-transgenic Mice. J. Biol. Chem. 2000, 275, 3693–3698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.Y.; Ju, S.M.; Lee, J.A.; Kwon, H.-J.; Eum, W.S.; Jang, S.H.; Choi, S.Y.; Park, J. Suppression of HIV-1 Tat-induced monocyte adhesiveness by a cell-permeable superoxide dismutase in astrocytes. Exp. Mol. Med. 2007, 39, 778–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, J.M.; Melov, S. SOD2 in mitochondrial dysfunction and neurodegeneration. Free Radic. Biol. Med. 2013, 62, 4–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisiger, R.A.; Fridovich, I. Mitochondrial superoxide simutase. Site of synthesis and intramitochondrial localization. J. Biol. Chem. 1973, 13, 4793–4796. [Google Scholar] [CrossRef]
- Lebovitz, R.M.; Zhang, H.; Vogel, H.; Cartwright, J., Jr.; Dionne, L.; Lu, N.; Huang, S.; Matzuk, M.M. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9782–9787. [Google Scholar] [CrossRef] [Green Version]
- Marcus, D.L.; AStrafaci, J.; Freedman, M.L. Differential neuronal expression of manganese superoxide dismutase in Alzheimer’s disease. Med. Sci. Monit. 2006, 12, BR8–BR14. [Google Scholar]
- Massaad, C.A.; Washington, T.M.; Pautler, R.G.; Klann, E. Overexpression of SOD-2 reduces hippocampal superoxide and prevents memory deficits in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 13576–13581. [Google Scholar] [CrossRef] [Green Version]
- Saha, R.N.; Pahan, K. Differential regulation of Mn-superoxide dismutase in neurons and astroglia by HIV-1 gp120: Implications for HIV-associated dementia. Free Radic. Biol. Med. 2007, 42, 1866–1878. [Google Scholar] [CrossRef] [Green Version]
- Moon, T.C.; Befus, A.D.; Kulka, M. Mast Cell Mediators: Their Differential Release and the Secretory Pathways Involved. Front. Immunol. 2014, 5, 569. [Google Scholar] [CrossRef] [Green Version]
- Luo, T.; Chen, B.; Zhao, Z.; He, N.; Zeng, Z.; Wu, B.; Fukushima, Y.; Dai, M.; Huang, Q.; Xu, D.; et al. Histamine H2 receptor activation exacerbates myocardial ischemia/reperfusion injury by disturbing mitochondrial and endothelial function. Basic Res. Cardiol. 2013, 108, 342. [Google Scholar] [CrossRef] [PubMed]
- Zidar, D.A.; Juchnowski, S.; Ferrari, B.; Clagett, B.; Pilch-Cooper, H.A.; Rose, S.; Rodriguez, B.; McComsey, G.A.; Sieg, S.F.; Mehta, N.; et al. Oxidized LDL Levels Are Increased in HIV Infection and May Drive Monocyte Activation. JAIDS J. Acquir. Immune Defic. Syndr. 2015, 69, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Teer, E.; Essop, M.F. HIV and Cardiovascular Disease: Role of Immunometabolic Perturbations. Physiology 2018, 33, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Fukami, K.; Yamagishi, S.-I.; Okuda, S. Role of AGEs-RAGE System in Cardiovascular Disease. Curr. Pharm. Des. 2014, 20, 2395–2402. [Google Scholar] [CrossRef]
- Fang, F.; Yu, Q.; Arancio, O.; Chen, D.; Gore, S.S.; Yan, S.S.; Yan, S.F. RAGE mediates Aβ accumulation in a mouse model of Alzheimer’s disease via modulation of β- and γ-secretase activity. Hum. Mol. Genet. 2018, 27, 1002–1014. [Google Scholar] [CrossRef] [Green Version]
- Loeser, R.F.; Yammani, R.R.; Carlson, C.S.; Chen, H.; Cole, A.; Im, H.-J.; Bursch, L.S.; Du Yan, S. Articular chondrocytes express the receptor for advanced glycation end products: Potential role in osteoarthritis. Arthritis Care Res. 2005, 52, 2376–2385. [Google Scholar] [CrossRef] [Green Version]
- Simm, A.; Caßelmann, C.; Schubert, A.; Hofmann, S.; Reimann, A.; Silber, R.-E. Age associated changes of AGE-receptor expression: RAGE upregulation is associated with human heart dysfunction. Exp. Gerontol. 2004, 39, 407–413. [Google Scholar] [CrossRef]
- Grossin, N.; Auger, F.; Niquet-Leridon, C.; Durieux, N.; Montaigne, D.; Schmidt, A.M.; Susen, S.; Jacolot, P.; Beuscart, J.-B.; Tessier, F.J.; et al. Dietary CML-enriched protein induces functional arterial aging in a RAGE-dependent manner in mice. Mol. Nutr. Food Res. 2015, 59, 927–938. [Google Scholar] [CrossRef]
- Mao, Y.X.; Cai, W.J.; Sun, X.Y.; Dai, P.P.; Li, X.M.; Wang, Q.; Huang, X.L.; He, B.; Wang, P.P.; Wu, G.; et al. RAGE-dependent mitochondria pathway: A novel target of silibinin against apoptosis of osteoblastic cells induced by advanced glycation end products. Cell Death Dis. 2018, 9, 674. [Google Scholar] [CrossRef] [Green Version]
- Lo, M.-C.; Chen, M.-H.; Lee, W.-S.; Lu, C.-I.; Chang, C.-R.; Kao, S.-H.; Lee, H.-M. Nε-(carboxymethyl) lysine-induced mitochondrial fission and mitophagy cause decreased insulin secretion from β-cells. Am. J. Physiol. Metab. 2015, 309, E829–E839. [Google Scholar] [CrossRef] [Green Version]
- Teissier, T.; Boulanger, É. The receptor for advanced glycation end-products (RAGE) is an important pattern recognition receptor (PRR) for inflammaging. Biogerontology 2019, 20, 279–301. [Google Scholar] [CrossRef] [PubMed]
- Reddy, G.K. AGE-related cross-linking of collagen is associated with aortic wall matrix stiffness in the pathogenesis of drug-induced diabetes in rats. Microvasc. Res. 2004, 68, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Dickens, A.M.; Yoo, S.W.; Chin, A.C.; Xu, J.; Johnson, T.P.; Trout, A.L.; Hauser, K.F.; Haughey, N.J. Chronic low-level expression of HIV-1 Tat promotes a neurodegenerative phenotype with aging. Sci. Rep. 2017, 7, 7748. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gender | Male | Female | ||||||
| Tat Genotype | Tat(−) | Tat(+) | Tat(−) | Tat(+) | Tat(−) | Tat(+) | Tat(−) | Tat(+) |
| n-value | 8 | 13 | 5 | 15 | 11 | 12 | 24 | 22 |
| Treatment | Saline | Dox 56 mg/kg | Saline | Dox 56 mg/kg | ||||
| HR (bpm) | 454.09 ± 49.21 | 488.37 ± 52.74 | 473.52 ± 45.52 | 477.33 ± 70.09 | 465.07 ± 88.05 | 465.07 ± 88.05 | 470.70 ± 53.07 | 483.51 ± 98.17 |
| B-mode | B-mode | |||||||
| SV (µL) | 18.21 ± 3.44 | 24.38 ± 4.82 | 20.04 ± 7.98 | 19.83 ± 5.45 | 17.69 ± 4.13 | 17.29 ± 3.24 | 19.75 ± 6.26 | 20.20 ± 5.63 |
| %EF | 48.66 ± 5.61 | 50.33 ± 9.22 | 49.71 ± 8.49 | 47.49 ± 9.67 | 44.52 ± 9.78 | 53.31 ± 11.05 | 52.02 ± 9.22 | 55.28 ± 7.76 |
| %FS | 15.10 ± 4.44 | 14.01 ± 5.81 | 13.11 ± 5.92 | 12.18 ± 4.20 | 10.11 ± 3.98 | 12.69 ± 4.22 | 13.45 ± 5.18 | 13.52 ± 5.76 |
| CO (mL/min) | 8.02 ± 1.59 | 11.73 ± 1.73 | 9.14 ± 2.77 | 9.40 ± 3.07 | 8.23 ± 2.70 | 8.31 ± 2.67 | 9.16 ± 2.77 | 9.89 ± 3.68 |
| Area (mm2) | 11.91 ± 3.67 | 14.47 ± 3.70 | 13.84 ± 5.62 | 12.75 ± 3.51 | 14.45 ± 4.01 | 9.98 ± 2.13 | 10.99 ± 2.00 | 11.73 ± 2.73 |
| Area, s (mm2) | 11.10 ± 2.32 | 13.39 ± 3.24 | 12.18 ± 4.27 | 12.33 ± 2.69 | 12.66 ± 2.35 | 9.66 ± 1.75 | 10.68 ± 1.82 | 10.15 ± 2.05 |
| Area, d (mm2) | 16.73 ± 2.54 | 20.25 ± 2.86 | 17.61 ± 5.13 | 18.16 ± 3.07 | 17.91 ± 2.48 | 15.20 ± 1.31 | 16.81 ± 2.62 | 16.39 ± 2.51 |
| Volume (µL) | 24.10 ± 12.14 | 30.55 + 11.56 | 29.94 ± 17.20 | 24.25 ± 10.25 | 30.18 ± 13.00 | 17.29 ± 6.68 | 18.38 ± 5.05 | 22.25 ± 9.68 |
| Volume, s (µL) | 20.41 ± 7.06 | 26.70 ± 10.18 | 23.74 ± 11.05 | 22.58 ± 7.45 | 23.48 ± 6.73 | 16.03 ± 5.51 | 17.48 ± 4.38 | 16.60 ± 5.12 |
| Volume, d (µL) | 38.62 ± 9.94 | 51.07 ± 11.39 | 20.04 ± 7.98 | 42.41 ± 10.55 | 41.17 ± 8.66 | 33.31 ± 5.44 | 37.24 ± 8.27 | 36.79 ± 9.44 |
| M-mode | M-mode | |||||||
| %EF | 63.15 ± 14.73 | 55.27 ± 16.51 | 55.42 ± 6.98 | 62.91 ± 10.61 | 61.30 ± 6.39 | 61.77 ± 11.27 | 63.48 ± 7.18 | 64.55 ± 6.62 |
| %FS | 34.77 ± 10.05 | 29.97 ± 11.94 | 28.46 ± 4.31 | 33.85 ± 7.51 | 32.28 ± 4.49 | 33.21 ± 7.07 | 33.70 ± 5.26 | 34.45 ± 4.78 |
| IVS, d (mm) | 1.08 ± 0.19 | 0.99 ± 0.23 | 0.75 ± 0.05 | 1.16 ± 0.26 | 0.89 ± 0.21 | 1.04 ± 0.24 | 1.13 ± 0.24 | 0.91 ± 0.20 |
| IVS, s (mm) | 1.48 ± 0.21 | 1.38 ± 0.38 | 1.07 ± 0.06 | 1.50 ± 0.28 | 1.30 ± 0.23 | 1.37 ± 0.21 | 1.56 ± 0.25 | 1.35 ± 0.25 |
| LVID, d (mm) | 3.27 ± 0.36 | 3.65 ± 0.59 | 3.65 ± 0.32 | 3.28 ± 0.39 | 3.36 ± 0.36 | 3.07 ± 0.30 | 3.09 ± 0.37 | 3.12 ± 0.37 |
| LVID, s (mm) | 2.15 ± 0.49 | 2.64 ± 0.78 | 2.62 ± 0.31 | 2.19 ± 0.38 | 2.28 ± 0.31 | 2.04 ± 0.27 | 2.06 ± 0.32 | 2.04 ± 0.32 |
| LVPW, d (mm) | 1.22 ± 0.27 | 1.01 ± 0.23 | 0.91 ± 0.16 | 1.08 ± 0.20 | 0.97 ± 0.31 | 1.02 ± 0.20 | 1.16 ± 0.31 | 1.09 ± 0.28 |
| LVPW, s (mm) | 1.57 ± 0.33 | 1.33 ± 0.25 | 1.29 ± 0.20 | 1.43 ± 0.23 | 1.30 ± 0.27 | 1.37 ± 0.20 | 1.46 ± 0.28 | 1.43 ± 0.25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qrareya, A.N.; Wise, N.S.; Hodges, E.R.; Mahdi, F.; Stewart, J.A., Jr.; Paris, J.J. HIV-1 Tat Upregulates the Receptor for Advanced Glycation End Products and Superoxide Dismutase-2 in the Heart of Transgenic Mice. Viruses 2022, 14, 2191. https://doi.org/10.3390/v14102191
Qrareya AN, Wise NS, Hodges ER, Mahdi F, Stewart JA Jr., Paris JJ. HIV-1 Tat Upregulates the Receptor for Advanced Glycation End Products and Superoxide Dismutase-2 in the Heart of Transgenic Mice. Viruses. 2022; 14(10):2191. https://doi.org/10.3390/v14102191
Chicago/Turabian StyleQrareya, Alaa N., Nason S. Wise, Emmanuel R. Hodges, Fakhri Mahdi, James A. Stewart, Jr., and Jason J. Paris. 2022. "HIV-1 Tat Upregulates the Receptor for Advanced Glycation End Products and Superoxide Dismutase-2 in the Heart of Transgenic Mice" Viruses 14, no. 10: 2191. https://doi.org/10.3390/v14102191