
Ampliseq for Illumina Technology Enables Detailed Molecular Epidemiology of Rabies Lyssaviruses from Infected Formalin-Fixed Paraffin-Embedded Tissues


Abstract
:1. Introduction
2. Materials and Methods
2.1. Rabies-Positive Unfixed Samples
2.2. Rabies-Positive FFPE Samples
2.3. RNA Extraction
2.4. RT-qPCR
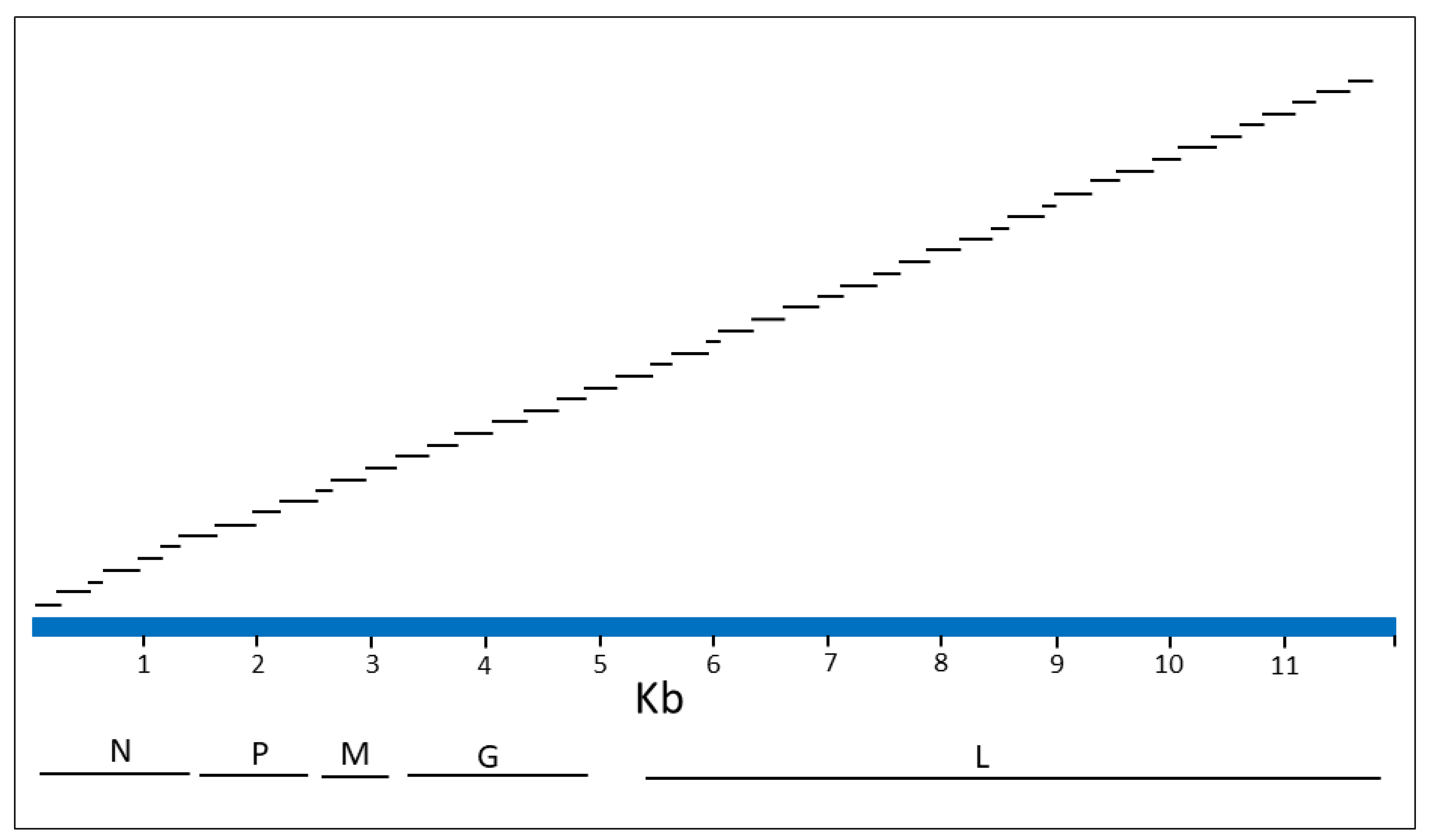
2.5. Design of Ampliseq for Illumina Panel
2.6. Ampliseq for Illumina Protocol
2.7. Species Composition of Illumina Reads
2.8. Reference-Guided Assembly and Viral Type Determinations
2.9. Phylogenetic Analysis
2.10. Statistical Analysis
3. Results
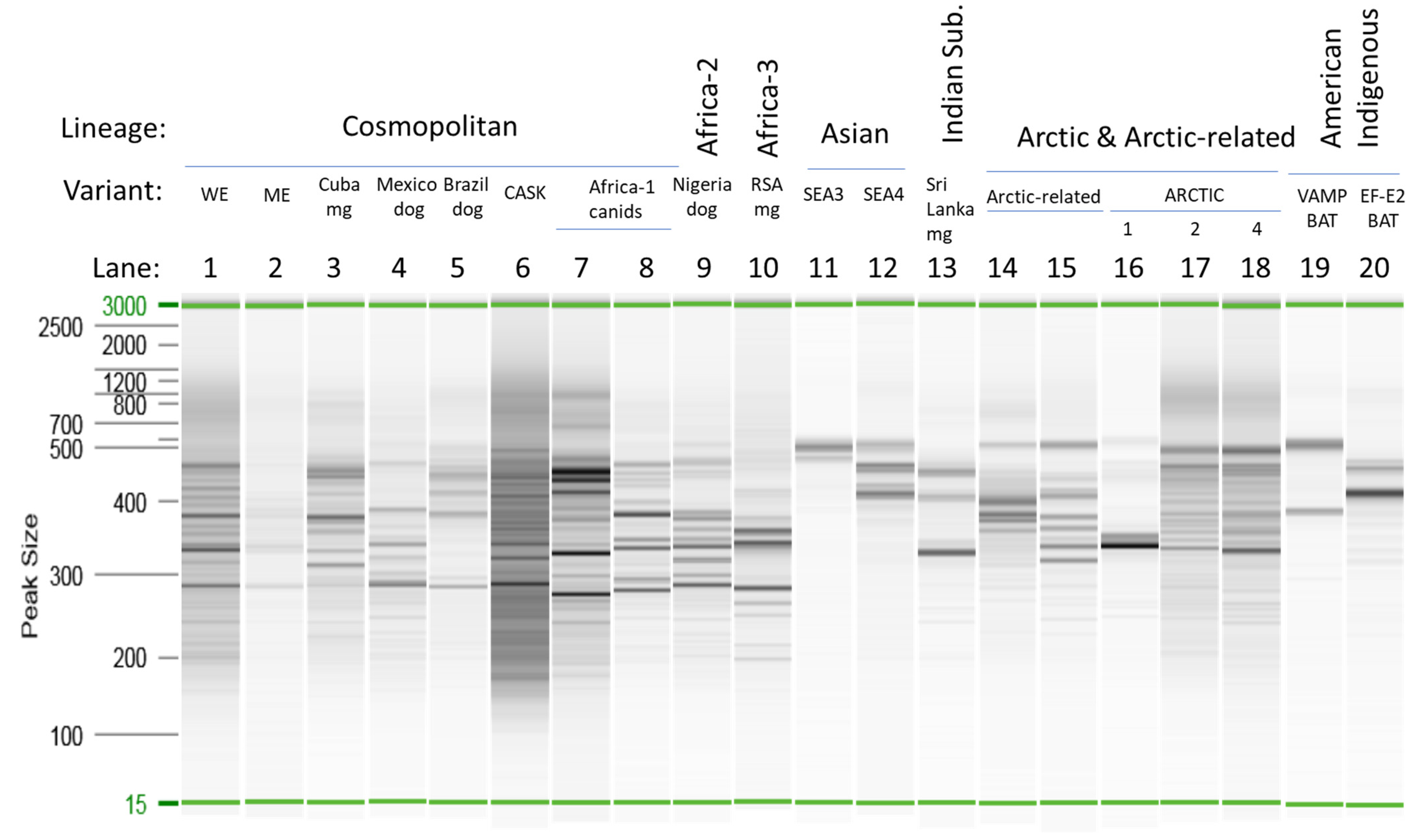
3.1. Amplicon Size Distribution Analysis
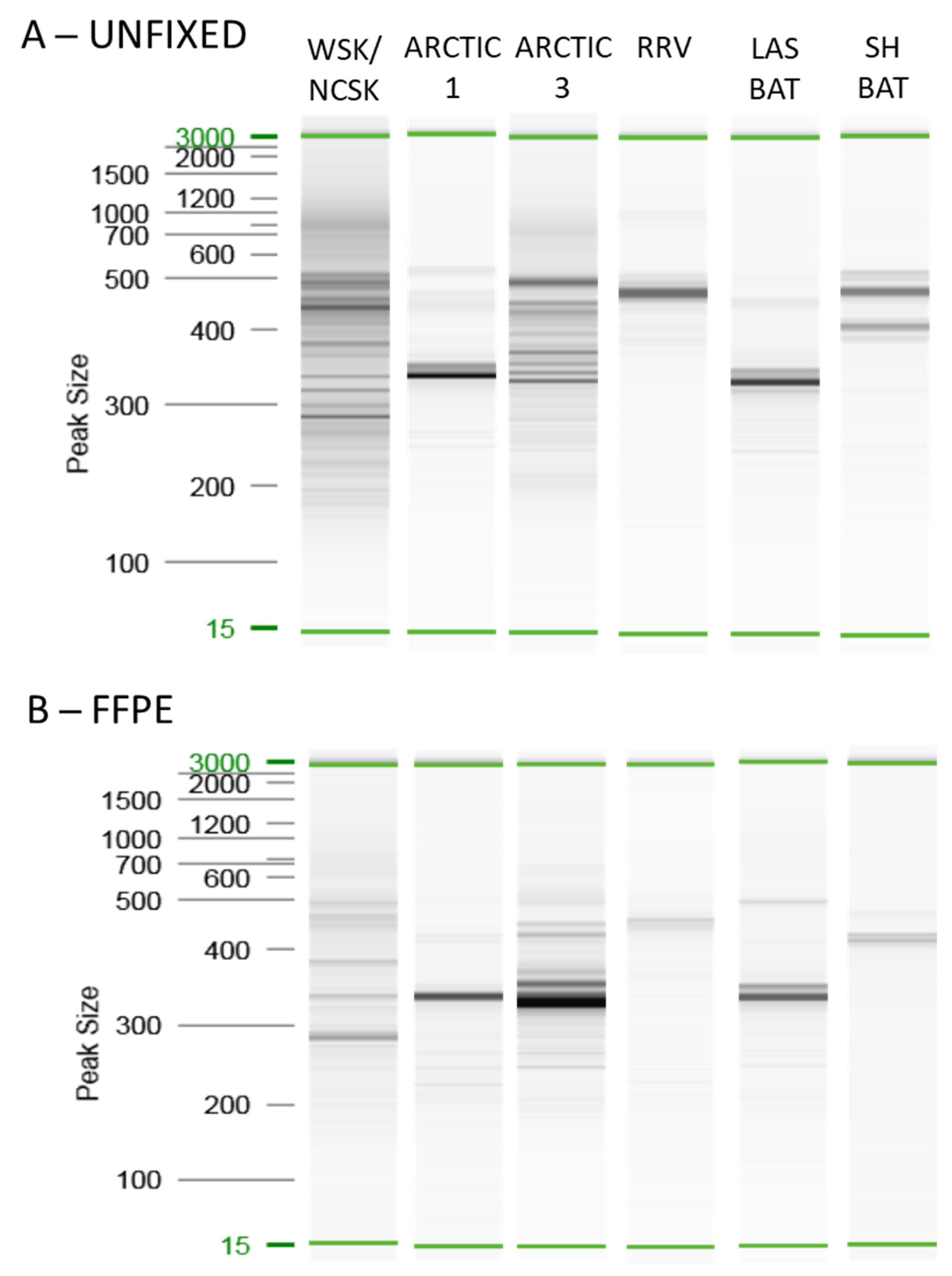
3.1.1. Unfixed Samples
3.1.2. Comparison of Unfixed and FFPE Samples
3.2. Species Assignment of Illumina Reads
3.3. Amplicon Sequence Analysis
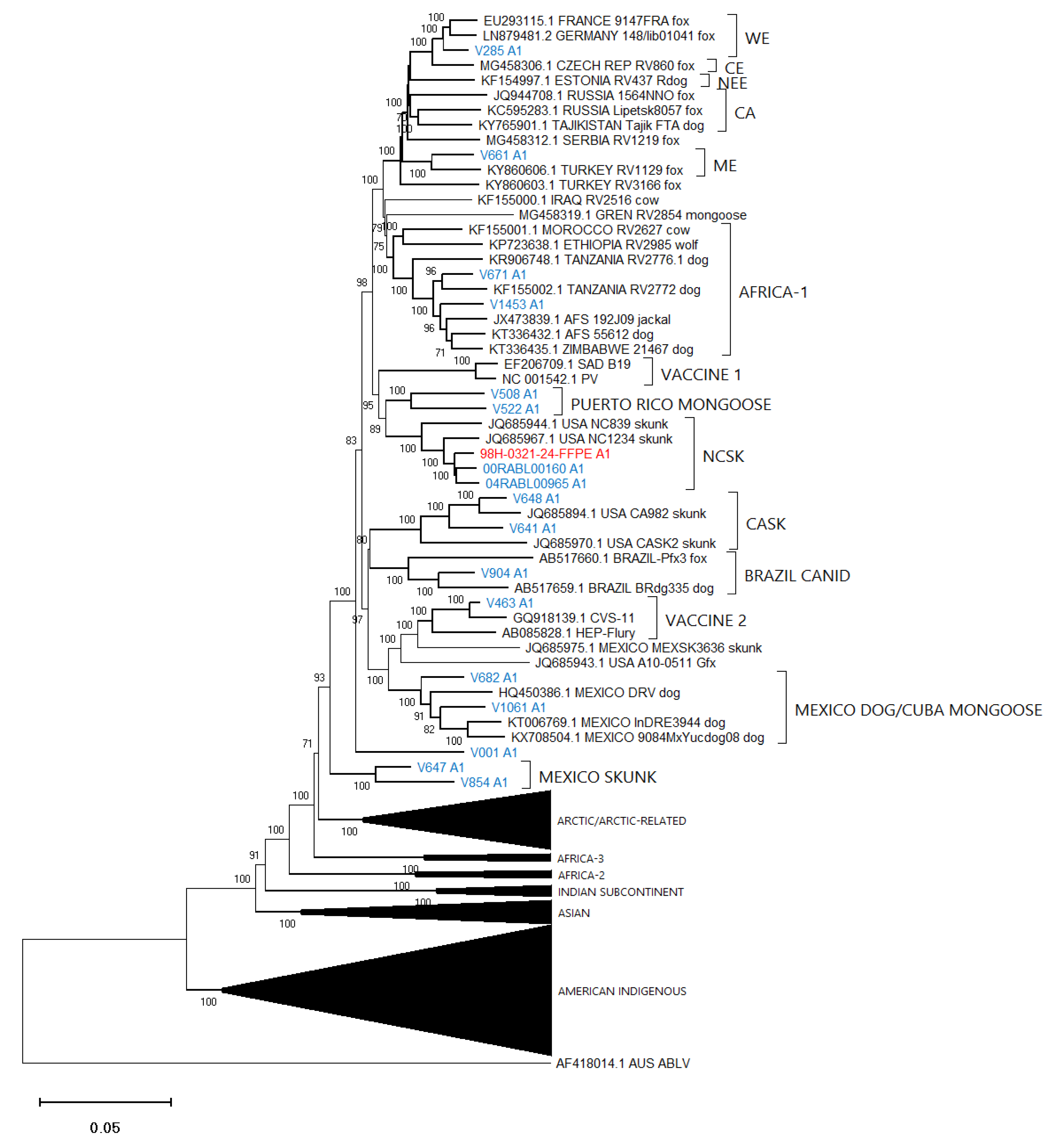
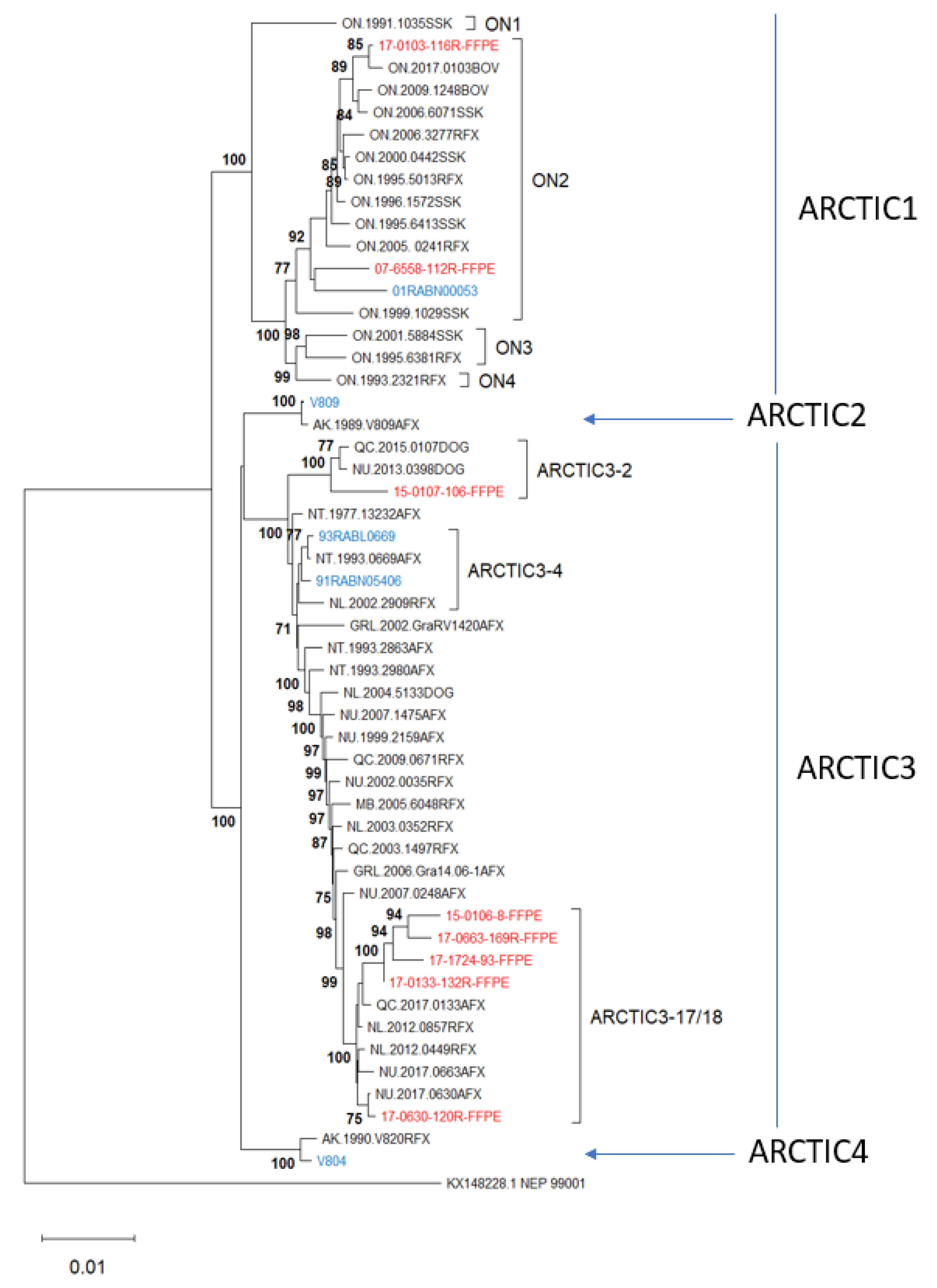
3.4. Phylogenetic Analysis
3.5. Optimization of Viral Variant Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dean, D.J.; Abelseth, M.K.; Atanasiu, P. The fluorescent antibody test. In Laboratory Techniques in Rabies, 4th ed.; Meslin, F.X., Kaplan, M.M., Koprowski, H., Eds.; World Health Organization (WHO): Geneva, Switzerland, 1996; pp. 88–95. [Google Scholar]
- Fehlner-Gardiner, C.; Nadin-Davis, S.; Armstrong, J.; Muldoon, F.; Bachmann, P.; Wandeler, A. ERA vaccine-derived cases of rabies in wildlife and domestic animals in Ontario, Canada, 1989–2004. J. Wildl. Dis. 2008, 44, 71–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.S.; Reid-Sanden, F.L.; Roumillat, L.F.; Trimarchi, C.; Clark, K.; Baer, G.M.; Winkler, W.G. Demonstration of antigenic variation among rabies virus isolates by using monoclonal antibodies to nucleocapsid proteins. J. Clin. Microbiol. 1986, 24, 573–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marston, D.A.; Jennings, D.L.; MacLaren, N.C.; Dorey-Robinson, D.; Fooks, A.R.; Banyard, A.C.; McElhinney, L.M. Pan-lyssavirus real time RT-PCR for rabies diagnosis. J. Vis. Exp. 2019, 149, e59709. [Google Scholar] [CrossRef] [Green Version]
- Wadhwa, A.; Wilkins, K.; Gao, J.; Condori Condori, R.E.; Gigante, C.M.; Zhao, H.; Ma, X.; Ellison, J.A.; Greenberg, L.; Velasco-Villa, A.; et al. A Pan-lyssavirus Taqman real-time RT-PCR assay for the detection of highly variable rabies virus and other lyssaviruses. PLOS Negl. Trop. Dis. 2017, 11, e0005258. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Orciari, L.A.; Yager, P.A.; Seidel, H.D.; Warner, C.K. Epidemiologic and historical relationships among 87 rabies virus isolates as determined by limited sequence analysis. J. Infect. Dis. 1992, 166, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Troupin, C.; Dacheux, L.; Tanguy, M.; Sabeta, C.; Blanc, H.; Bouchier, C.; Vignuzzi, M.; Duchene, S.; Holmes, E.C.; Bourhy, H. Large-scale phylogenomic analysis reveals the complex evolutionary history of rabies virus in multiple carnivore hosts. PLoS Pathog. 2016, 12, e1006041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunker, K.; Marston, D.A.; Horton, D.L.; Cleaveland, S.; Fooks, A.R.; Kazwala, R.; Ngeleja, C.; Lembo, T.; Sambo, M.; Mtema, Z.J.; et al. Elucidating the phylodynamics of endemic rabies virus in eastern Africa using whole-genome sequencing. Virus Evol. 2015, 1, vev011. [Google Scholar] [CrossRef] [Green Version]
- Trewby, H.; Nadin-Davis, S.A.; Real, L.A.; Biek, R. Processes underlying rabies virus incursions across US-Canada border as revealed by whole-genome phylogeography. Emerg. Infect. Dis. 2017, 23, 1454–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balachandran, A.; Charlton, K. Experimental rabies infection of non-nervous tissues in skunks (Mephitis mephitis) and foxes (Vulpes vulpes). Vet. Pathol. 1994, 31, 93–102. [Google Scholar] [CrossRef] [Green Version]
- Niezgoda, M.; Satheshkumar, P.S. Immunohistochemistry test for the lyssavirus antigen detection from formalin-fixed tissues. J. Vis. Exp. 2021, 176, 60138. [Google Scholar] [CrossRef]
- Warner, C.K.; Whitfield, S.G.; Fekadu, M.; Ho, H. Procedures for reproducible detection of rabies virus antigen mRNA and genome in situ in formalin-fixed tissues. J. Virol. Methods 1997, 67, 5–12. [Google Scholar] [CrossRef]
- Nadin-Davis, S.A.; Sheen, M.; Wandeler, A.I. Use of discriminatory probes for strain typing of formalin-fixed, rabies virus-infected tissues by in situ hybridization. J. Clin. Microbiol. 2003, 41, 4343–4352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wacharapluesadee, S.; Ruangvejvorachai, P.; Hemachudha, T. A simple method for detection of rabies viral sequences in 16-year old archival brain specimens with one-week fixation in formalin. J. Virol. Methods 2006, 134, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Condori, R.E.; Niezgoda, M.; Lopez, G.; Matos, C.A.; Mateo, E.D.; Gigante, C.; Hartloge, C.; Filpo, A.P.; Haim, J.; Satheshkumar, P.S.; et al. Using the LN34 pan-lyssavirus real-time RT-PCR assay for rabies diagnosis and rapid genetic typing from formalin-fixed human brain tissue. Viruses 2020, 12, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanke, D.; Freuling, C.M.; Fischer, S.; Hueffer, K.; Hundertmark, K.; Nadin-Davis, S.; Marston, D.; Fooks, A.R.; Botner, A.; Mettenleiter, T.C.; et al. Saptio-temporal analysis of the genetic diversity of Arctic rabies viruses and their reservoir hosts in Greenland. PLoS Negl. Trop. Dis. 2016, 10, e0004779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, K.M.; Eshleman, J.R. Simultaneous sequencing of multiple polymerase chain reaction products and combined polymerase chain reaction with cycle sequencing in single reactions. Am. J. Pathol. 2002, 161, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Towler, W.I.; Church, J.D.; Eshleman, J.R.; Fowler, M.G.; Guay, L.A.; Jackson, J.B.; Eshleman, S.H. Analysis of nevirapine resistance mutations in cloned HIV Type 1 variants from HIV-Infected Ugandan infants using a single-step amplification-sequencing method (AmpliSeq). AIDS Res. Hum. Retrovir. 2008, 24, 1209–1213. [Google Scholar] [CrossRef] [PubMed]
- Alessandrini, F.; Caucci, S.; Onofri, V.; Melchionda, F.; Tagliabracci, A.; Bagnarelli, P.; Di Sante, L.; Turchi, C.; Menzo, S. Evaluation of the Ion AmpliSeq SARS-CoV-2 research panel by massive parallel sequencing. Genes 2020, 11, 929. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, A.K.; Selli, C.; Martinez-Perez, C.; Fernando, A.; Renshaw, L.; Keys, J.; Figueroa, J.D.; He, X.; Tanioka, M.; Munro, A.F.; et al. Unlocking the transcriptomic potential of formalin-fixed paraffin embedded clinical tissues: Comparison of gene expression profiling approaches. BMC Bioinform. 2020, 21, 30. [Google Scholar] [CrossRef] [PubMed]
- Buechler, S.A.; Stephens, M.T.; Hummon, A.B.; Ludwig, K.; Cannon, E.; Carter, T.C.; Resnick, J.; Gökmen-Polar, Y.; Badve, S.S. ColoType: A forty gene signature for consensus molecular subtyping of colorectal cancer tumors using whole-genome assay or targeted RNA-sequencing. Sci. Rep. 2020, 10, 12123. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, L.; Sah, S.; Latham, G.J.; Patel, R.; Song, Q.; Koeppen, H.; Tam, R.; Schleifman, E.; Mashhedi, H.; et al. Profiling cancer gene mutations in clinical formalin-fixed, paraffin-embedded colorectal tumor specimens using targeted Next-Generation Sequencing. Oncology 2014, 19, 336–343. [Google Scholar] [CrossRef] [Green Version]
- Nadin-Davis, S.; Alnabelseya, N.; Knowles, M.K. The phylogeography of Myotis bat-associated rabies viruses across Canada. PLoS Negl. Trop. Dis. 2017, 11, e0005541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadin-Davis, S.A.; Feng, Y.; Mousse, D.; Wandeler, A.I.; Aris-Brosou, S. Spatial and temporal dynamics of rabies virus variants in big brown bat populations across Canada: Footprints of an emerging zoonosis. Mol. Ecol. 2010, 19, 2120–2136. [Google Scholar] [CrossRef] [PubMed]
- Nadin-Davis, S.A.; Huang, W.; Armstrong, J.; Casey, G.A.; Bahloul, C.; Tordo, N.; Wandeler, A.I. Antigenic and genetic divergence of rabies viruses from bat species indigenous to Canada. Virus Res. 2001, 74, 139–156. [Google Scholar] [CrossRef]
- Koprowski, H. The mouse inoculation test. In Laboratory Techniques in Rabies, 4th ed.; Meslin, F.X., Kaplan, M.M., Koprowski, H., Eds.; World Health Organization: Geneva, Switzerland, 1996; pp. 80–87. [Google Scholar]
- Nadin-Davis, S.A.; Sheen, M.; Wandeler, A.I. Development of real-time reverse transcriptase polymerase chain reaction methods for human rabies diagnosis. J. Med. Virol. 2009, 81, 1484–1497. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- Breitwieser, F.P.; Salzberg, S.L. Pavian: Interactive analysis of metagenomics data for microbiome studies and pathogen identification. Bioinformatics 2020, 36, 1303–1304. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [Green Version]
- Davis, R.; Nadin-Davis, S.A.; Moore, M.; Hanlon, C. Genetic characterization and phylogenetic analysis of skunk-associated rabies viruses in North America with special emphasis on the central plains. Virus Res. 2013, 174, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Nadin-Davis, S.A.; Velez, J.; Malaga, C.; Wandeler, A.I. A molecular epidemiological study of rabies in Puerto Rico. Virus Res. 2008, 131, 8–15. [Google Scholar] [CrossRef]
- Nadin-Davis, S.A.; Abdel-Malik, M.; Armstrong, J.; Wandeler, A.I. Lyssavirus P gene characterisation provides insights into the phylogeny of the genus and identifies structural similarities and diversity within the encoded phosphoprotein. Virology 2002, 298, 286–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuzmin, I.V.; Shi, M.; Orciari, L.A.; Yager, P.A.; Velasco-Villa, A.; Kuzmina, N.A.; Streicker, D.G.; Bergman, D.L.; Rupprecht, C.E. Molecular inferences suggest multiple host shifts of rabies viruses from bats to mesocarniovores in Arizona during 2001–2009. PLoS Pathog. 2012, 8, e1002786. [Google Scholar] [CrossRef] [PubMed]
- Nadin-Davis, S.A.; Falardeau, E.; Flynn, A.; Whitney, H.; Marshall, H.D. Relationships between fox populations and rabies virus spread in northern Canada. PLoS ONE 2021, 16, e0246508. [Google Scholar] [CrossRef] [PubMed]
- Nadin-Davis, S.A.; Fehlner-Gardiner, C. Origins of the arctic fox variant rabies viruses responsible for recent cases of the disease in southern Ontario. PLOS Negl. Trop. Dis. 2019, 13, e0007699. [Google Scholar] [CrossRef] [PubMed]
- Nadin-Davis, S.A.; Casey, G.A.; Wandeler, A. Identification of regional variants of the rabies virus within the Canadian province of Ontario. J. Gen. Virol. 1993, 74, 829–837. [Google Scholar] [CrossRef]
- Le Mercier, P.; Jacob, Y.; Tordo, N. The complete Mokola virus genome sequence: Structure of the RNA-dependent RNA polymerase. J. Gen. Virol. 1997, 78, 1571–1576. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | 01RABN00053 | 72RABL03675 | V1145 | V1375 | V804 | V809 | V982 |
|---|---|---|---|---|---|---|---|
| Host species | Mephitis mephitis | Eptesicus fuscus | Canis familiaris | Canis familiaris | Vulpes vulpes | Vulpes lagopus | Bos taurus |
| Number of raw reads | 225,275 | 433,412 | 201,030 | 224,329 | 254,338 | 312,591 | 416,666 |
| Classified reads (%) | 99.55 | 99.40 | 98.98 | 98.20 | 99.26 | 99.49 | 98.46 |
| Unclassified reads (%) | 0.45 | 0.60 | 1.02 | 1.80 | 0.74 | 0.51 | 1.54 |
| Viral reads (%) | 99.49 | 99.38 | 98.08 | 97.85 | 99.24 | 99.49 | 98.42 |
| Chordate reads (%) | 0.06 | 0.02 | 0.91 | 0.35 | 0.01 | 0.00 | 0.04 |
| Canidae reads (%) | 0.00 | 0.00 | 0.87 | 0.19 | 0.00 | 0.00 | 0.01 |
| Mephitidae reads (%) | 0.04 | 0.00 | 0.02 | 0.01 | 0.00 | 0.00 | 0.00 |
| Bovidae reads (%) | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.01 |
| Chiroptera reads (%) | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Comments | 99 reads assigned to Mephitidae | 4 reads assigned to Eptesicus fuscus | 1746 reads assigned to Canidae | 424 reads assigned to Canidae | 8 reads assigned to Vulpes sp. | 3 reads assigned to Vulpes sp. | 21 reads assigned to Bos taurus |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nadin-Davis, S.A.; Hartke, A.; Kang, M. Ampliseq for Illumina Technology Enables Detailed Molecular Epidemiology of Rabies Lyssaviruses from Infected Formalin-Fixed Paraffin-Embedded Tissues. Viruses 2022, 14, 2241. https://doi.org/10.3390/v14102241
Nadin-Davis SA, Hartke A, Kang M. Ampliseq for Illumina Technology Enables Detailed Molecular Epidemiology of Rabies Lyssaviruses from Infected Formalin-Fixed Paraffin-Embedded Tissues. Viruses. 2022; 14(10):2241. https://doi.org/10.3390/v14102241
Chicago/Turabian StyleNadin-Davis, Susan Angela, Allison Hartke, and Mingsong Kang. 2022. "Ampliseq for Illumina Technology Enables Detailed Molecular Epidemiology of Rabies Lyssaviruses from Infected Formalin-Fixed Paraffin-Embedded Tissues" Viruses 14, no. 10: 2241. https://doi.org/10.3390/v14102241