
Diversity and Ecology of Caudoviricetes Phages with Genome Terminal Repeats in Fecal Metagenomes from Four Dutch Cohorts

 , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus Detection in Metagenomes
2.2. Nucleotide Sequence Characterization
2.3. Identification of Potential Plasmids
2.4. Genetic Code Prediction
2.5. Proteome Annotation
2.6. Taxonomic Assignment Based on Marker Genes
2.7. Species-Level Clustering
2.8. Read Mapping
2.9. Building TerL MSA
2.10. Building the Phylogenetic Tree
2.11. The Gene-Sharing Network Reconstruction
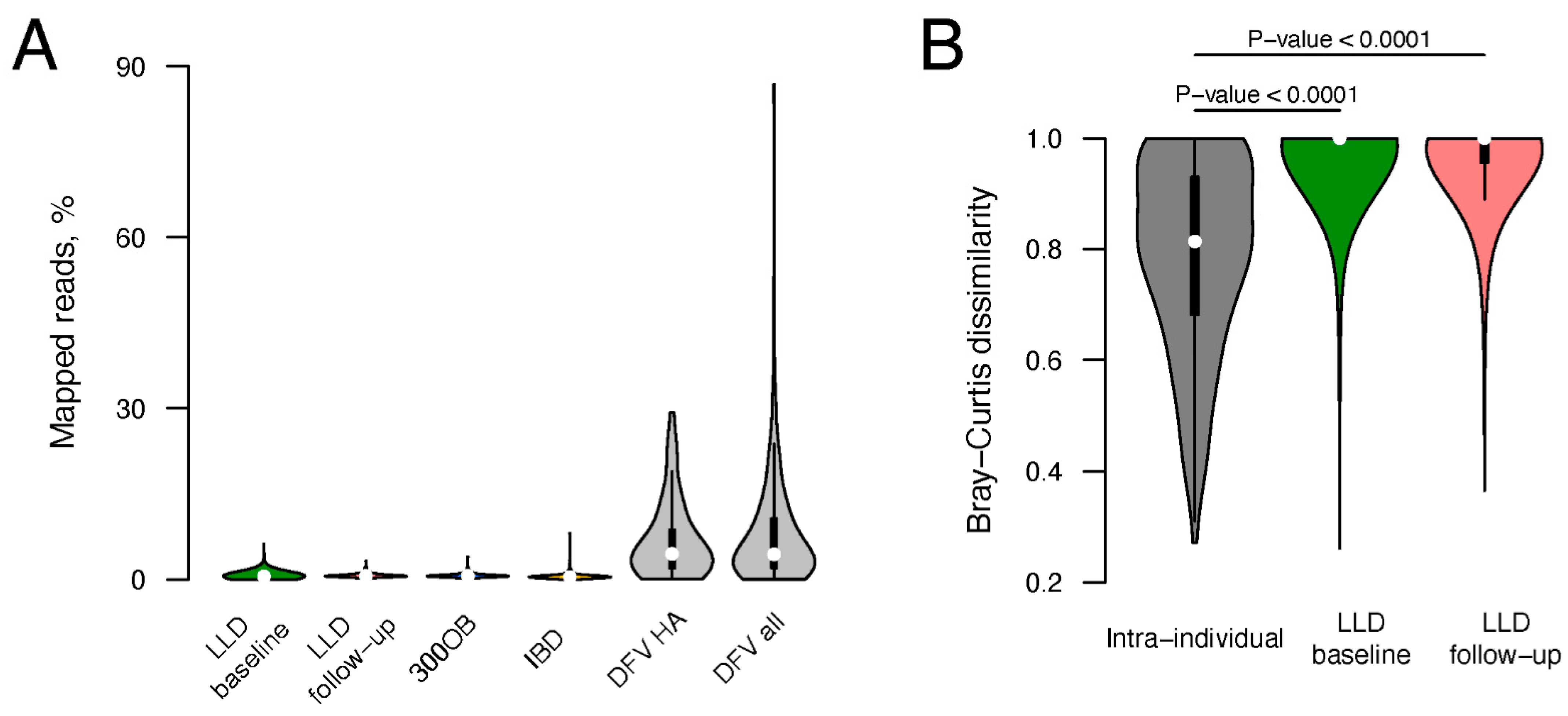
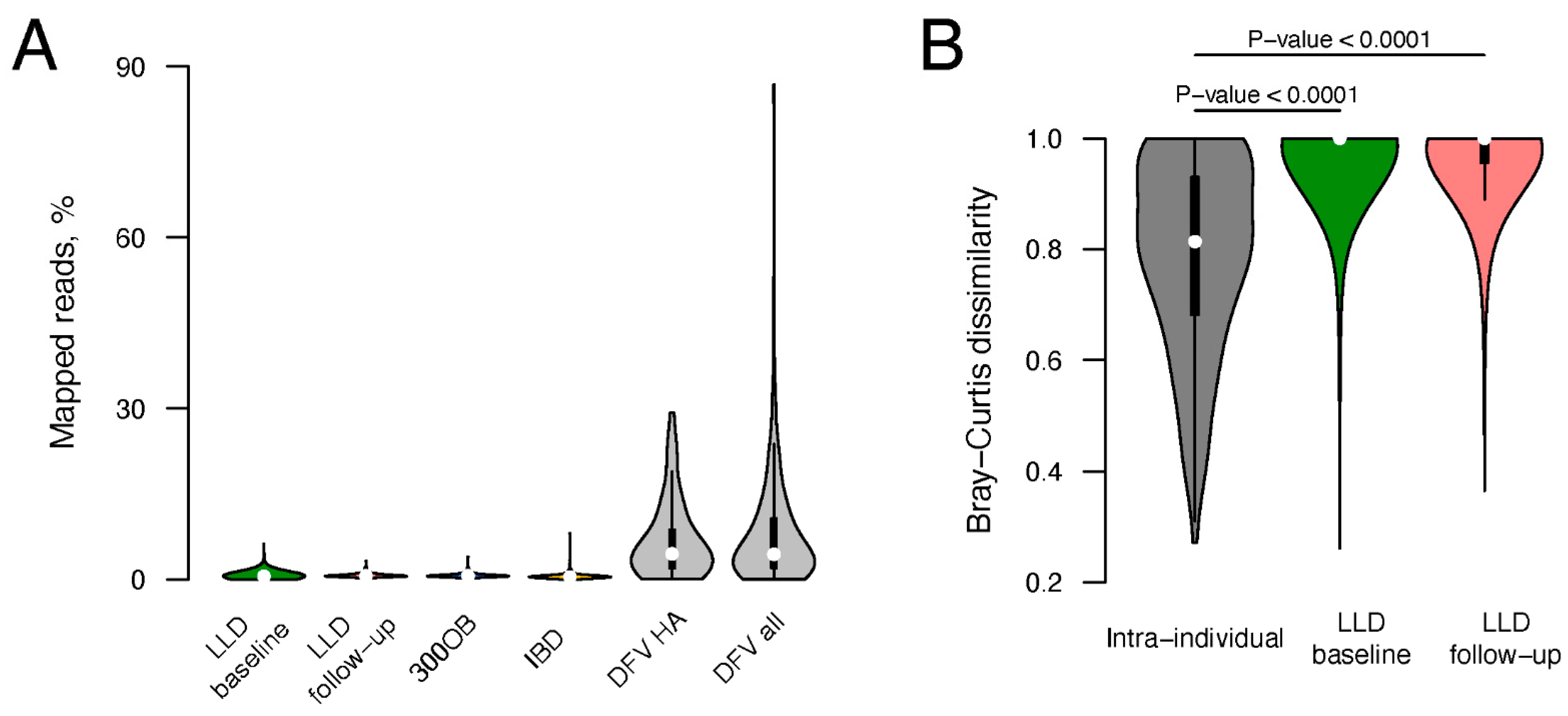
2.12. Virome Stability Estimation
2.13. Prophage-Based Host Prediction
2.14. CRISPR-Based Host Prediction
2.15. Co-Abundance-Based Host Prediction
2.16. Finding Similar Extensively Characterized Phages
2.17. Associations with Human Phenotypes
2.18. Visualization
3. Results
3.1. Viral Fraction of Total Fecal Metagenomes
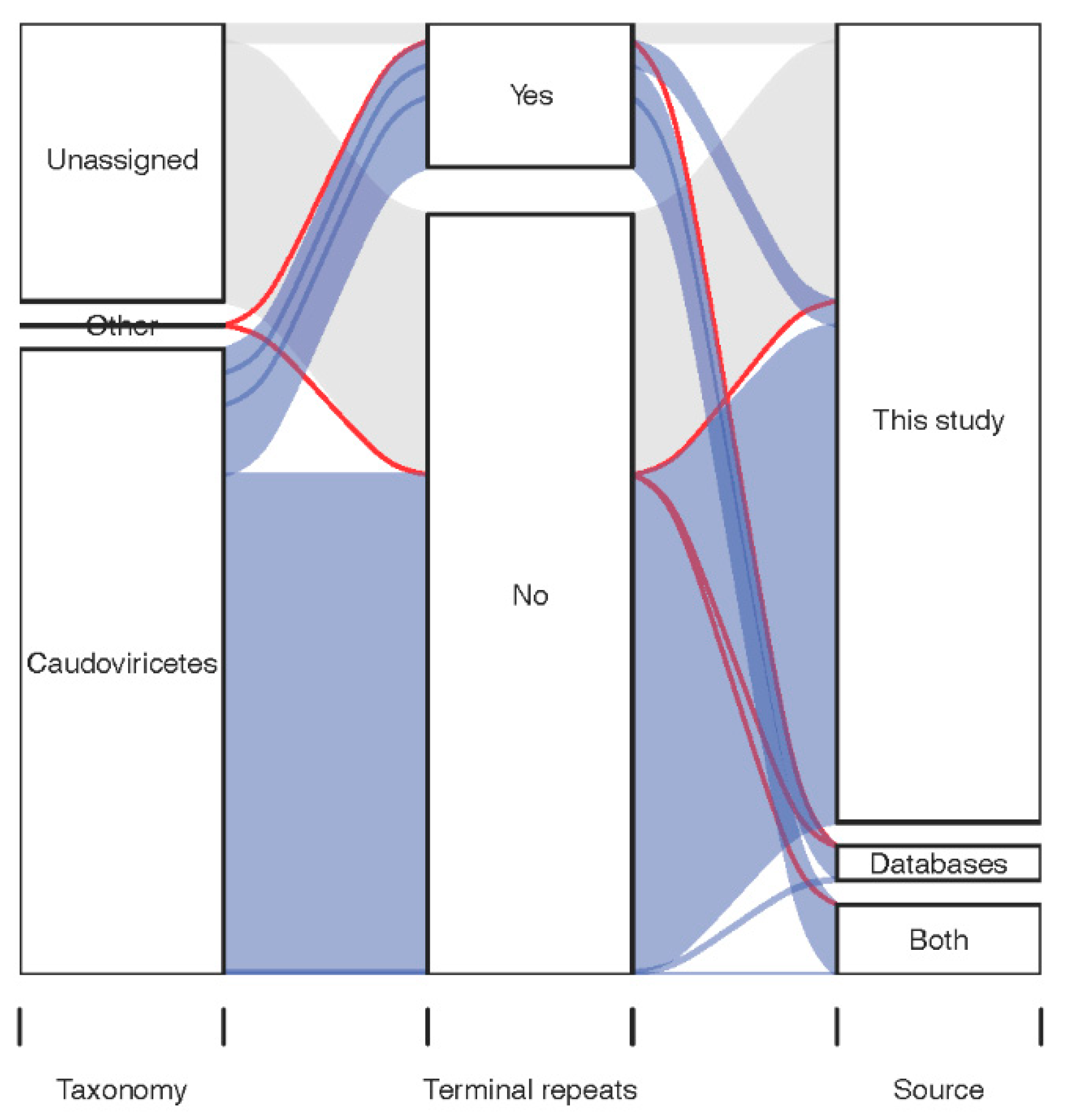
3.2. Diversity of Caudoviricetes Phages with Genome Terminal Repeats
3.3. Diversity of the Most Prevalent Caudoviricetes Phages with Genome Terminal Repeats

3.4. Associations with Human Phenotypes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koonin, E.V.; Dolja, V.V.; Krupovic, M. The healthy human virome: From virus-host symbiosis to disease. Curr. Opin. Virol. 2021, 47, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Benler, S.; Koonin, E.V. Fishing for phages in metagenomes: What do we catch, what do we miss? Curr. Opin. Virol. 2021, 49, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Nayfach, S.; Paez-Espino, D.; Call, L.; Low, S.J.; Sberro, H.; Ivanova, N.N.; Proal, A.D.; Fischbach, M.A.; Bhatt, A.S.; Hugenholtz, P.; et al. Metagenomic compendium of 189,680 DNA viruses from the human gut microbiome. Nat. Microbiol. 2021, 6, 960–970. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Bushman, F.D. The human virome: Assembly, composition and host interactions. Nat. Rev. Microbiol. 2021, 19, 514–527. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A Roadmap for Genome-Based Phage Taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef] [PubMed]
- Iranzo, J.; Krupovic, M.; Koonin, E.V. The Double-Stranded DNA Virosphere as a Modular Hierarchical Network of Gene Sharing. mBio 2016, 7, e00978-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adriaenssens, E.M. Phage Diversity in the Human Gut Microbiome: A Taxonomist’s Perspective. mSystems 2021, 6, e0079921. [Google Scholar] [CrossRef] [PubMed]
- Casjens, S.R.; Gilcrease, E.B. Determining DNA packaging strategy by analysis of the termini of the chromosomes in tailed-bacteriophage virions. Methods Mol. Biol. 2009, 502, 91–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merrill, B.D.; Ward, A.T.; Grose, J.H.; Hope, S. Software-based analysis of bacteriophage genomes, physical ends, and packaging strategies. BMC Genom. 2016, 17, 679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meijer, W.J.; Horcajadas, J.A.; Salas, M. Phi29 family of phages. Microbiol. Mol. Biol. Rev. 2001, 65, 261–287. [Google Scholar] [CrossRef] [PubMed]
- Kieft, K.; Anantharaman, K. Virus genomics: What is being overlooked? Curr. Opin. Virol. 2022, 53, 101200. [Google Scholar] [CrossRef] [PubMed]
- Mantynen, S.; Laanto, E.; Oksanen, H.M.; Poranen, M.M.; Diaz-Munoz, S.L. Black box of phage-bacterium interactions: Exploring alternative phage infection strategies. Open Biol. 2021, 11, 210188. [Google Scholar] [CrossRef] [PubMed]
- Howard-Varona, C.; Hargreaves, K.R.; Abedon, S.T.; Sullivan, M.B. Lysogeny in nature: Mechanisms, impact and ecology of temperate phages. ISME J. 2017, 11, 1511–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Dempsey, D.M.; Dutilh, B.E.; Garcia, M.L.; Curtis Hendrickson, R.; et al. Recent changes to virus taxonomy ratified by the International Committee on Taxonomy of Viruses (2022). Arch. Virol. 2022, 167, 2429–2440. [Google Scholar] [CrossRef] [PubMed]
- Garmaeva, S.; Sinha, T.; Kurilshikov, A.; Fu, J.; Wijmenga, C.; Zhernakova, A. Studying the gut virome in the metagenomic era: Challenges and perspectives. BMC Biol. 2019, 17, 84. [Google Scholar] [CrossRef]
- Tigchelaar, E.F.; Zhernakova, A.; Dekens, J.A.; Hermes, G.; Baranska, A.; Mujagic, Z.; Swertz, M.A.; Munoz, A.M.; Deelen, P.; Cenit, M.C.; et al. Cohort profile: LifeLines DEEP, a prospective, general population cohort study in the northern Netherlands: Study design and baseline characteristics. BMJ Open 2015, 5, e006772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhernakova, A.; Kurilshikov, A.; Bonder, M.J.; Tigchelaar, E.F.; Schirmer, M.; Vatanen, T.; Mujagic, Z.; Vila, A.V.; Falony, G.; Vieira-Silva, S.; et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 2016, 352, 565–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Wang, D.; Garmaeva, S.; Kurilshikov, A.; Vich Vila, A.; Gacesa, R.; Sinha, T.; Lifelines Cohort, S.; Segal, E.; Weersma, R.K.; et al. The long-term genetic stability and individual specificity of the human gut microbiome. Cell 2021, 184, 2302–2315.e12. [Google Scholar] [CrossRef]
- Ter Horst, R.; van den Munckhof, I.C.L.; Schraa, K.; Aguirre-Gamboa, R.; Jaeger, M.; Smeekens, S.P.; Brand, T.; Lemmers, H.; Dijkstra, H.; Galesloot, T.E.; et al. Sex-Specific Regulation of Inflammation and Metabolic Syndrome in Obesity. Arter. Thromb. Vasc Biol. 2020, 40, 1787–1800. [Google Scholar] [CrossRef] [PubMed]
- Kurilshikov, A.; van den Munckhof, I.C.L.; Chen, L.; Bonder, M.J.; Schraa, K.; Rutten, J.H.W.; Riksen, N.P.; de Graaf, J.; Oosting, M.; Sanna, S.; et al. Gut Microbial Associations to Plasma Metabolites Linked to Cardiovascular Phenotypes and Risk. Circ. Res. 2019, 124, 1808–1820. [Google Scholar] [CrossRef]
- Vich Vila, A.; Imhann, F.; Collij, V.; Jankipersadsing, S.A.; Gurry, T.; Mujagic, Z.; Kurilshikov, A.; Bonder, M.J.; Jiang, X.; Tigchelaar, E.F.; et al. Gut microbiota composition and functional changes in inflammatory bowel disease and irritable bowel syndrome. Sci. Transl. Med. 2018, 10, eaap8914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imhann, F.; Van der Velde, K.J.; Barbieri, R.; Alberts, R.; Voskuil, M.D.; Vich Vila, A.; Collij, V.; Spekhorst, L.M.; Van der Sloot, K.W.J.; Peters, V.; et al. The 1000IBD project: Multi-omics data of 1000 inflammatory bowel disease patients; data release 1. BMC Gastroenterol. 2019, 19, 5. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulyaeva, A.; Garmaeva, S.; Ruigrok, R.; Wang, D.; Riksen, N.P.; Netea, M.G.; Wijmenga, C.; Weersma, R.K.; Fu, J.; Vila, A.V.; et al. Discovery, diversity, and functional associations of crAss-like phages in human gut metagenomes from four Dutch cohorts. Cell Rep. 2022, 38, 110204. [Google Scholar] [CrossRef]
- Tisza, M.J.; Belford, A.K.; Dominguez-Huerta, G.; Bolduc, B.; Buck, C.B. Cenote-Taker 2 democratizes virus discovery and sequence annotation. Virus Evol. 2021, 7, veaa100. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.K.; Fogarty, E.C.; Eren, A.M. The genetic and ecological landscape of plasmids in the human gut. bioRxiv 2022. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanova, N.N.; Schwientek, P.; Tripp, H.J.; Rinke, C.; Pati, A.; Huntemann, M.; Visel, A.; Woyke, T.; Kyrpides, N.C.; Rubin, E.M. Stop codon reassignments in the wild. Science 2014, 344, 909–913. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.; Huber, W.; Pages, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef] [PubMed]
- Charif, D.; Lobry, J.R. SeqinR 1.0-2: A Contributed Package to the R Project for Statistical Computing Devoted to Biological Sequences Retrieval and Analysis. In Structural Approaches to Sequence Evolution: Molecules, Networks, Populations; Bastolla, U., Porto, M., Roman, H.E., Vendruscolo, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 207–232. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Benler, S.; Yutin, N.; Antipov, D.; Rayko, M.; Shmakov, S.; Gussow, A.B.; Pevzner, P.; Koonin, E.V. Thousands of previously unknown phages discovered in whole-community human gut metagenomes. Microbiome 2021, 9, 78. [Google Scholar] [CrossRef]
- Yutin, N.; Benler, S.; Shmakov, S.A.; Wolf, Y.I.; Tolstoy, I.; Rayko, M.; Antipov, D.; Pevzner, P.A.; Koonin, E.V. Analysis of metagenome-assembled viral genomes from the human gut reveals diverse putative CrAss-like phages with unique genomic features. Nat. Commun. 2021, 12, 1044. [Google Scholar] [CrossRef]
- Grazziotin, A.L.; Koonin, E.V.; Kristensen, D.M. Prokaryotic Virus Orthologous Groups (pVOGs): A resource for comparative genomics and protein family annotation. Nucleic Acids Res. 2017, 45, D491–D498. [Google Scholar] [CrossRef]
- Yutin, N.; Backstrom, D.; Ettema, T.J.G.; Krupovic, M.; Koonin, E.V. Vast diversity of prokaryotic virus genomes encoding double jelly-roll major capsid proteins uncovered by genomic and metagenomic sequence analysis. Virol. J. 2018, 15, 67. [Google Scholar] [CrossRef]
- Aylward, F.O.; Moniruzzaman, M.; Ha, A.D.; Koonin, E.V. A phylogenomic framework for charting the diversity and evolution of giant viruses. PLoS Biol. 2021, 19, e3001430. [Google Scholar] [CrossRef]
- Wheeler, T.J.; Clements, J.; Finn, R.D. Skylign: A tool for creating informative, interactive logos representing sequence alignments and profile hidden Markov models. BMC Bioinform. 2014, 15, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aylward, F.O.; Moniruzzaman, M. ViralRecall-A Flexible Command-Line Tool for the Detection of Giant Virus Signatures in ‘Omic Data. Viruses 2021, 13, 150. [Google Scholar] [CrossRef] [PubMed]
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 2020, 39, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Adriaenssens, E.M.; Dutilh, B.E.; Koonin, E.V.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Lavigne, R.; Brister, J.R.; Varsani, A.; et al. Minimum Information about an Uncultivated Virus Genome (MIUViG). Nat. Biotechnol. 2019, 37, 29–37. [Google Scholar] [CrossRef]
- Van Espen, L.; Bak, E.G.; Beller, L.; Close, L.; Deboutte, W.; Juel, H.B.; Nielsen, T.; Sinar, D.; De Coninck, L.; Frithioff-Bojsoe, C.; et al. A Previously Undescribed Highly Prevalent Phage Identified in a Danish Enteric Virome Catalog. mSystems 2021, 6, e0038221. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Roux, S.; Emerson, J.B.; Eloe-Fadrosh, E.A.; Sullivan, M.B. Benchmarking viromics: An in silico evaluation of metagenome-enabled estimates of viral community composition and diversity. PeerJ 2017, 5, e3817. [Google Scholar] [CrossRef] [Green Version]
- Steinegger, M.; Meier, M.; Mirdita, M.; Vohringer, H.; Haunsberger, S.J.; Soding, J. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinform. 2019, 20, 473. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, B.J.; Rodrigues, A.P.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Whelan, S.; Goldman, N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 2001, 18, 691–699. [Google Scholar] [CrossRef] [Green Version]
- Schliep, K.P. phangorn: Phylogenetic analysis in R. Bioinformatics 2011, 27, 592–593. [Google Scholar] [CrossRef] [Green Version]
- Gaïa, M.; Meng, L.; Pelletier, E.; Forterre, P.; Vanni, C.; Fernandez-Guerra, A.; Jaillon, O.; Wincker, P.; Ogata, H.; Krupovic, M.; et al. Plankton-infecting relatives of herpesviruses clarify the evolutionary trajectory of giant viruses. bioRxiv 2022. [Google Scholar] [CrossRef]
- Bin Jang, H.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Pruitt, K.D.; Schoch, C.L.; Sherry, S.T.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2022, 50, D161–D164. [Google Scholar] [CrossRef]
- Shmakov, S.A.; Sitnik, V.; Makarova, K.S.; Wolf, Y.I.; Severinov, K.V.; Koonin, E.V. The CRISPR Spacer Space Is Dominated by Sequences from Species-Specific Mobilomes. mBio 2017, 8, e01397-17. [Google Scholar] [CrossRef]
- Pourcel, C.; Touchon, M.; Villeriot, N.; Vernadet, J.P.; Couvin, D.; Toffano-Nioche, C.; Vergnaud, G. CRISPRCasdb a successor of CRISPRdb containing CRISPR arrays and cas genes from complete genome sequences, and tools to download and query lists of repeats and spacers. Nucleic Acids Res. 2020, 48, D535–D544. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Paez-Espino, D.; Chen, I.A.; Palaniappan, K.; Ratner, A.; Chu, K.; Reddy, T.B.K.; Nayfach, S.; Schulz, F.; Call, L.; et al. IMG/VR v3: An integrated ecological and evolutionary framework for interrogating genomes of uncultivated viruses. Nucleic Acids Res. 2021, 49, D764–D775. [Google Scholar] [CrossRef] [PubMed]
- Beghini, F.; McIver, L.J.; Blanco-Miguez, A.; Dubois, L.; Asnicar, F.; Maharjan, S.; Mailyan, A.; Manghi, P.; Scholz, M.; Thomas, A.M.; et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. Elife 2021, 10, e65088. [Google Scholar] [CrossRef] [PubMed]
- Balduzzi, S.; Rücker, G.; Schwarzer, G. How to perform a meta-analysis with R: A practical tutorial. Evid. Based Ment. Health 2019, 22, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Society. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Wagih, O. ggseqlogo: A versatile R package for drawing sequence logos. Bioinformatics 2017, 33, 3645–3647. [Google Scholar] [CrossRef] [Green Version]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [Green Version]
- Brister, J.R.; Ako-Adjei, D.; Bao, Y.; Blinkova, O. NCBI viral genomes resource. Nucleic Acids Res. 2015, 43, D571–D577. [Google Scholar] [CrossRef] [Green Version]
- Soto-Perez, P.; Bisanz, J.E.; Berry, J.D.; Lam, K.N.; Bondy-Denomy, J.; Turnbaugh, P.J. CRISPR-Cas System of a Prevalent Human Gut Bacterium Reveals Hyper-targeting against Phages in a Human Virome Catalog. Cell Host Microbe 2019, 26, 325–335.e325. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.C.; Zablocki, O.; Zayed, A.A.; Howell, A.; Bolduc, B.; Sullivan, M.B. The Gut Virome Database Reveals Age-Dependent Patterns of Virome Diversity in the Human Gut. Cell Host Microbe 2020, 28, 724–740.e8. [Google Scholar] [CrossRef] [PubMed]
- Camarillo-Guerrero, L.F.; Almeida, A.; Rangel-Pineros, G.; Finn, R.D.; Lawley, T.D. Massive expansion of human gut bacteriophage diversity. Cell 2021, 184, 1098–1109.e1099. [Google Scholar] [CrossRef] [PubMed]
- Devoto, A.E.; Santini, J.M.; Olm, M.R.; Anantharaman, K.; Munk, P.; Tung, J.; Archie, E.A.; Turnbaugh, P.J.; Seed, K.D.; Blekhman, R.; et al. Megaphages infect Prevotella and variants are widespread in gut microbiomes. Nat. Microbiol. 2019, 4, 693–700. [Google Scholar] [CrossRef] [Green Version]
- Al-Shayeb, B.; Sachdeva, R.; Chen, L.X.; Ward, F.; Munk, P.; Devoto, A.; Castelle, C.J.; Olm, M.R.; Bouma-Gregson, K.; Amano, Y.; et al. Clades of huge phages from across Earth’s ecosystems. Nature 2020, 578, 425–431. [Google Scholar] [CrossRef] [Green Version]
- Borges, A.L.; Lou, Y.C.; Sachdeva, R.; Al-Shayeb, B.; Penev, P.I.; Jaffe, A.L.; Lei, S.; Santini, J.M.; Banfield, J.F. Widespread stop-codon recoding in bacteriophages may regulate translation of lytic genes. Nat. Microbiol. 2022, 7, 918–927. [Google Scholar] [CrossRef]
- Kot, W.; Hammer, K.; Neve, H.; Vogensen, F.K. Identification of the receptor-binding protein in lytic Leuconostoc pseudomesenteroides bacteriophages. Appl. Env. Microbiol. 2013, 79, 3311–3314. [Google Scholar] [CrossRef] [Green Version]
- Kot, W.; Hansen, L.H.; Neve, H.; Hammer, K.; Jacobsen, S.; Pedersen, P.D.; Sorensen, S.J.; Heller, K.J.; Vogensen, F.K. Sequence and comparative analysis of Leuconostoc dairy bacteriophages. Int. J. Food Microbiol. 2014, 176, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Grigoriev, A. Analyzing genomes with cumulative skew diagrams. Nucleic Acids Res. 1998, 26, 2286–2290. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Deora, R.; Doulatov, S.R.; Gingery, M.; Eiserling, F.A.; Preston, A.; Maskell, D.J.; Simons, R.W.; Cotter, P.A.; Parkhill, J.; et al. Reverse transcriptase-mediated tropism switching in Bordetella bacteriophage. Science 2002, 295, 2091–2094. [Google Scholar] [CrossRef]
- Dutilh, B.E.; Cassman, N.; McNair, K.; Sanchez, S.E.; Silva, G.G.; Boling, L.; Barr, J.J.; Speth, D.R.; Seguritan, V.; Aziz, R.K.; et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 2014, 5, 4498. [Google Scholar] [CrossRef] [PubMed]
- Guerin, E.; Shkoporov, A.; Stockdale, S.R.; Clooney, A.G.; Ryan, F.J.; Sutton, T.D.S.; Draper, L.A.; Gonzalez-Tortuero, E.; Ross, R.P.; Hill, C. Biology and Taxonomy of crAss-like Bacteriophages, the Most Abundant Virus in the Human Gut. Cell Host Microbe 2018, 24, 653–664.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornuault, J.K.; Petit, M.A.; Mariadassou, M.; Benevides, L.; Moncaut, E.; Langella, P.; Sokol, H.; De Paepe, M. Phages infecting Faecalibacterium prausnitzii belong to novel viral genera that help to decipher intestinal viromes. Microbiome 2018, 6, 65. [Google Scholar] [CrossRef] [PubMed]
- Minot, S.; Grunberg, S.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Hypervariable loci in the human gut virome. Proc. Natl Acad Sci. USA 2012, 109, 3962–3966. [Google Scholar] [CrossRef] [Green Version]
- Dzunkova, M.; Low, S.J.; Daly, J.N.; Deng, L.; Rinke, C.; Hugenholtz, P. Defining the human gut host-phage network through single-cell viral tagging. Nat. Microbiol. 2019, 4, 2192–2203. [Google Scholar] [CrossRef] [PubMed]
- Ly, M.; Jones, M.B.; Abeles, S.R.; Santiago-Rodriguez, T.M.; Gao, J.; Chan, I.C.; Ghose, C.; Pride, D.T. Transmission of viruses via our microbiomes. Microbiome 2016, 4, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfennig, A.; Lomsadze, A.; Borodovsky, M. Annotation of Phage Genomes with Multiple Genetic Codes. bioRxiv 2022. [Google Scholar] [CrossRef]
- Weinheimer, A.R.; Aylward, F.O. Infection strategy and biogeography distinguish cosmopolitan groups of marine jumbo bacteriophages. ISME J. 2022, 16, 1657–1667. [Google Scholar] [CrossRef] [PubMed]
- Christie, G.E.; Dokland, T. Pirates of the Caudovirales. Virology 2012, 434, 210–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, H.; Saha, M.; Reyes-Aldrete, E.; Sherman, M.B.; Woodson, M.; Atz, R.; Grimes, S.; Jardine, P.J.; Morais, M.C. Structural and Molecular Basis for Coordination in a Viral DNA Packaging Motor. Cell Rep. 2016, 14, 2017–2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikkonen, M.; Alatossava, T. A group I intron in the terminase gene of Lactobacillus delbrueckii subsp. lactis phage LL-H. Microbiology 1995, 141 Pt 9, 2183–2190. [Google Scholar] [CrossRef] [PubMed]
- Bartolomucci, A.; Possenti, R.; Mahata, S.K.; Fischer-Colbrie, R.; Loh, Y.P.; Salton, S.R. The extended granin family: Structure, function, and biomedical implications. Endocr. Rev. 2011, 32, 755–797. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxonomic Group | Viral RefSeq Genome Sequences Recognized by Cenote-Taker 2, % | Taxonomic Assignment | ||
|---|---|---|---|---|
| Marker Protein Profile(s) b | Sensitivity, % | Specificity, % d | ||
| Class Caudoviricetes | 99.61 | Terminase large subunit (TerL): VOG00461 and alignments from Yutin et al., 2021 [38] and Benler et al., 2021 [37]. Target proteins were required to include hits to the TerL Walker B motif and the TerL nuclease motifs I and II. Target genomes encoding the Herpesviridae marker protein were discarded. | 94.29 | 99.98 |
| Family Herpesviridae | 82.46 | Major capsid protein (MCP): PF03122 | 79.82 | 100 |
| Family Papillomaviridae | 0 | Capsid protein L1: VOG05075 | 99.52 | 100 |
| Family Polyomaviridae | 0.76 | Coat protein: PF00718 | 99.24 | 100 |
| Family Adenoviridae | 91.89 | Hexon protein: VOG05391 | 91.89 | 100 |
| Class Tectiliviricetes (excluding the Adenoviridae family) | 89.47 | Double Jelly Roll MCP alignments corresponding to six prokaryotic virus groups c from Yutin et al., 2018 [40] | 100 | 99.99 |
| Phylum Nucleocytoviricota | 74.04 | Seven marker protein alignments from Aylward et al., 2021 [41]; each genome hit by a marker protein profile was further analyzed using ViralRecall (see Section 2) | 70.19 | 99.98 |
| Database | Viral Genomes Included | |
|---|---|---|
| Number | Description | |
| Viral RefSeq 209 | 6049 | Genome sequences ≥ 3000 nt, realm Riboviria excluded. |
| Human Virome Database (HuVirDB) | 1660 | Genome sequences ≥ 5000 nt with terminal repeats belonging a to the class Caudoviricetes. |
| Gut Virome Database (GVD) | 936 | |
| Gut Phage Database (GPD) | 10,870 | |
| Metagenomic Gut Virus (MGV) catalog | 19,694 | |
| Danish Enteric Virome Catalog (DEVoC) | 137 | |
| Devoto et al., 2019 [76] Al-Shayeb et al., 2020 [77] Borges et al., 2022 b [78] | 458 | |
| Benler et al., 2021 [37] | 1480 | Genome sequences belonging to the phylum Uroviricota. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gulyaeva, A.; Garmaeva, S.; Kurilshikov, A.; Vich Vila, A.; Riksen, N.P.; Netea, M.G.; Weersma, R.K.; Fu, J.; Zhernakova, A. Diversity and Ecology of Caudoviricetes Phages with Genome Terminal Repeats in Fecal Metagenomes from Four Dutch Cohorts. Viruses 2022, 14, 2305. https://doi.org/10.3390/v14102305
Gulyaeva A, Garmaeva S, Kurilshikov A, Vich Vila A, Riksen NP, Netea MG, Weersma RK, Fu J, Zhernakova A. Diversity and Ecology of Caudoviricetes Phages with Genome Terminal Repeats in Fecal Metagenomes from Four Dutch Cohorts. Viruses. 2022; 14(10):2305. https://doi.org/10.3390/v14102305
Chicago/Turabian StyleGulyaeva, Anastasia, Sanzhima Garmaeva, Alexander Kurilshikov, Arnau Vich Vila, Niels P. Riksen, Mihai G. Netea, Rinse K. Weersma, Jingyuan Fu, and Alexandra Zhernakova. 2022. "Diversity and Ecology of Caudoviricetes Phages with Genome Terminal Repeats in Fecal Metagenomes from Four Dutch Cohorts" Viruses 14, no. 10: 2305. https://doi.org/10.3390/v14102305
APA StyleGulyaeva, A., Garmaeva, S., Kurilshikov, A., Vich Vila, A., Riksen, N. P., Netea, M. G., Weersma, R. K., Fu, J., & Zhernakova, A. (2022). Diversity and Ecology of Caudoviricetes Phages with Genome Terminal Repeats in Fecal Metagenomes from Four Dutch Cohorts. Viruses, 14(10), 2305. https://doi.org/10.3390/v14102305