
Borna Disease Virus 1 Phosphoprotein Forms a Tetramer and Interacts with Host Factors Involved in DNA Double-Strand Break Repair and mRNA Processing
 , , , , , , , and
, , , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cloning and Plasmid Preparation
2.2. Protein Expression and Protein Purification
2.3. Crystallisation and X-ray Structure Determination
2.4. Small-Angle X-ray Scattering (SAXS) Analysis
2.5. Ab Initio Molecular Shape Analysis of POD
2.6. Full Atom Modelling of BoDV-1 PFULL
2.7. Minireplicon Assays
2.8. Determination of Proximal Host–Viral Protein Interactions in Living Cells (BioID)
3. Results
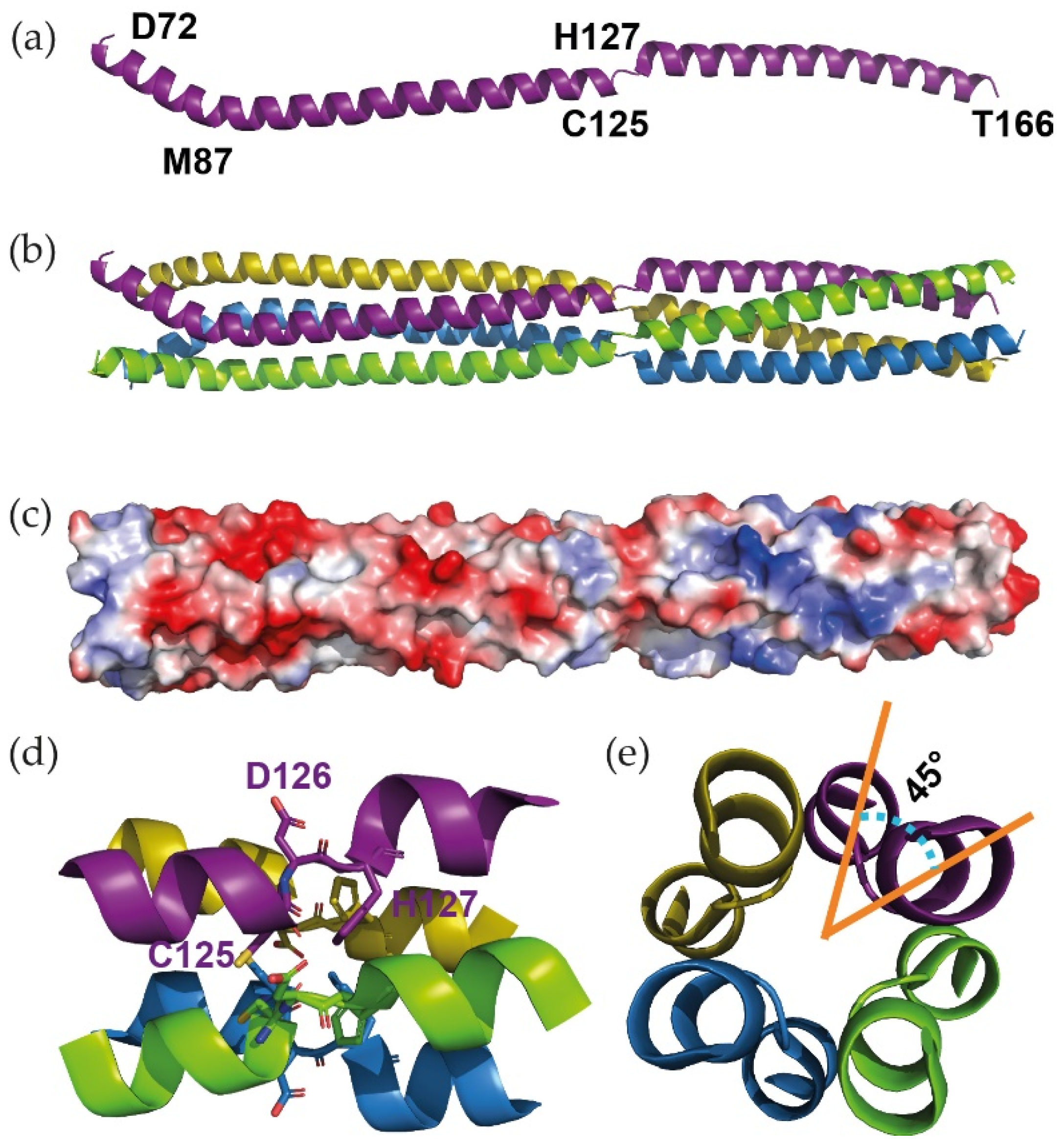
3.1. X-ray Structure of the Oligomerisation Domain of BoDV-1 Phosphoprotein
3.2. The β-Strand Twist of BoDV-1 P Is Important for Viral Replication
3.3. The β-Strand Twist Motif Is Conserved in Orthobornavirus POD
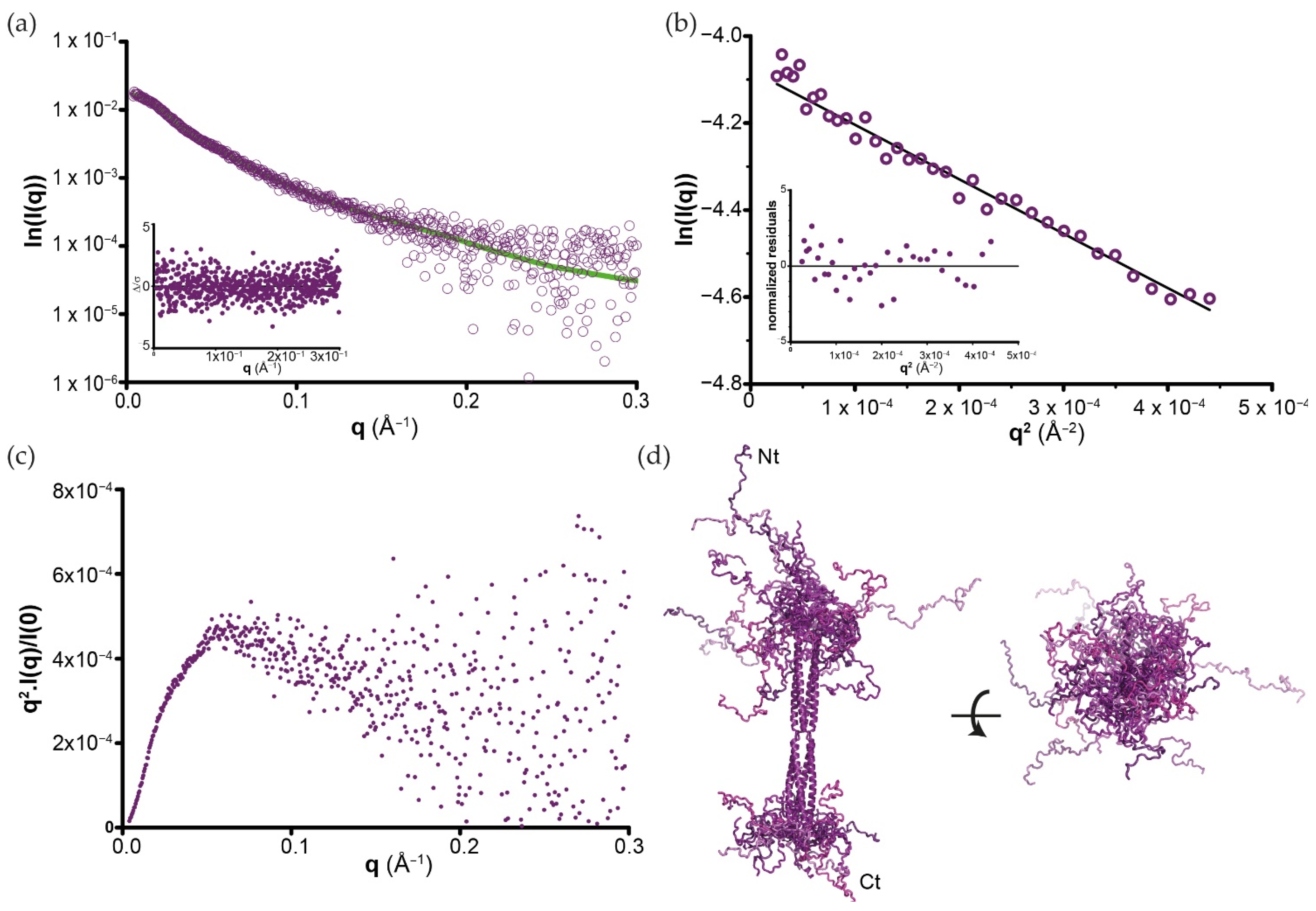
3.4. SAXS Analysis of Full-Length BoDV-1 Phosphoprotein
3.5. Determination of BoDV-1 P–Host Proximal Interactomics in Living Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Whelan, S.P.; Barr, J.N.; Wertz, G.W. Transcription and replication of nonsegmented negative-strand RNA viruses. Curr. Top. Microbiol. Immunol. 2004, 283, 61–119. [Google Scholar]
- Kolakofsky, D.; Le Mercier, P.; Nishio, M.; Blackledge, M.; Crepin, T.; Ruigrok, R.W.H. Sendai virus and a unified model of mononegavirus RNA synthesis. Viruses 2021, 13, 2466. [Google Scholar] [CrossRef]
- Tarbouriech, N.; Curran, J.; Ruigrok, R.W.; Burmeister, W.P. Tetrameric coiled-coil domain of Sendai virus phosphoprotein. Nat. Struct. Biol. 2000, 7, 777–781. [Google Scholar]
- Ding, H.; Green, T.J.; Lu, S.; Luo, M. Crystal structure of the oligomerization domain of the phosphoprotein of vesicular stomatitis virus. J. Virol. 2006, 80, 2808–2814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, I.; Crépin, T.; Jamin, M.; Ruigrok, R.W. Structure of the dimerization domain of the rabies virus phosphoprotein. J. Virol. 2010, 84, 3707–3710. [Google Scholar] [CrossRef] [Green Version]
- Communie, G.; Crépin, T.; Maurin, D.; Jensen, M.R.; Blackledge, M.; Ruigrok, R.W. Structure of the tetramerization domain of measles virus phosphoprotein. J. Virol. 2013, 87, 7166–7169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, R.; Green, T.J.; Purushotham, S.; Deivanayagam, C.; Bedwell, G.J.; Prevelige, P.E.; Luo, M. Structural and functional characterization of the mumps virus phosphoprotein. J. Virol. 2013, 87, 7558–7568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruhn, J.F.; Hotard, A.L.; Spiropoulou, C.F.; Lo, M.K.; Saphire, E.O. A conserved basic patch and central kink in the Nipah virus phosphoprotein multimerization domain are essential for polymerase function. Structure 2019, 27, 660–668 e4. [Google Scholar] [CrossRef] [Green Version]
- Zinzula, L.; Nagy, I.; Orsini, M.; Weyher-Stingl, E.; Bracher, A.; Baumeister, W. Structures of Ebola and Reston virus VP35 oligomerization domains and comparative biophysical characterization in all ebolavirus species. Structure 2019, 27, 39–54 e6. [Google Scholar] [CrossRef] [Green Version]
- Jensen, M.R.; Yabukarski, F.; Communie, G.; Condamine, E.; Mas, C.; Volchkova, V.; Tarbouriech, N.; Bourhis, J.M.; Volchkov, V.; Blackledge, M.; et al. Structural description of the Nipah virus phosphoprotein and its interaction with STAT1. Biophys. J. 2020, 118, 2470–2488. [Google Scholar] [CrossRef]
- Wang, Z.X.; Liu, S.B.; Guan, H.; Lu, L.F.; Tu, J.G.; Ouyang, S.; Zhang, Y.A. Structural and functional characterization of the phosphoprotein central domain of spring viremia of carp virus. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Cardone, C.; Caseau, C.M.; Bardiaux, B.; Thureaux, A.; Galloux, M.; Bajorek, M.; Eleouet, J.F.; Litaudon, M.; Bontems, F.; Sizun, C. A structural and dynamic analysis of the partially disordered polymerase-binding domain in RSV phosphoprotein. Biomolecules 2021, 11, 1225. [Google Scholar] [CrossRef] [PubMed]
- Gilman, M.S.A.; Liu, C.; Fung, A.; Behera, I.; Jordan, P.; Rigaux, P.; Ysebaert, N.; Tcherniuk, S.; Sourimant, J.; Eleouet, J.F.; et al. Structure of the respiratory syncytial virus polymerase complex. Cell 2019, 179, 193–204.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, D.; Gao, Y.; Roesler, C.; Rice, S.; D’Cunha, P.; Zhuang, L.; Slack, J.; Domke, M.; Antonova, A.; Romanelli, S.; et al. Cryo-EM structure of the respiratory syncytial virus RNA polymerase. Nat. Commun. 2020, 11, 368. [Google Scholar] [CrossRef] [Green Version]
- Abdella, R.; Aggarwal, M.; Okura, T.; Lamb, R.A.; He, Y. Structure of a paramyxovirus polymerase complex reveals a unique methyltransferase-CTD conformation. Proc. Natl. Acad. Sci. USA 2020, 117, 4931–4941. [Google Scholar] [CrossRef]
- Jenni, S.; Bloyet, L.M.; Diaz-Avalos, R.; Liang, B.; Whelan, S.P.J.; Grigorieff, N.; Harrison, S.C. Structure of the vesicular stomatitis virus L protein in complex with its phosphoprotein cofactor. Cell Rep. 2020, 30, 53–60.e5. [Google Scholar] [CrossRef] [Green Version]
- Horwitz, J.A.; Jenni, S.; Harrison, S.C.; Whelan, S.P.J. Structure of a rabies virus polymerase complex from electron cryo-microscopy. Proc. Natl. Acad. Sci. USA 2020, 117, 2099–2107. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Qian, X.; Lattmann, S.; El Sahili, A.; Yeo, T.H.; Jia, H.; Cressey, T.; Ludeke, B.; Noton, S.; Kalocsay, M.; et al. Structure of the human metapneumovirus polymerase phosphoprotein complex. Nature 2020, 577, 275–279. [Google Scholar] [CrossRef]
- Gould, J.R.; Qiu, S.; Shang, Q.; Ogino, T.; Prevelige, P.E., Jr.; Petit, C.M.; Green, T.J. The connector domain of vesicular stomatitis virus large protein interacts with the viral phosphoprotein. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Green, T.J.; Luo, M. Structure of the vesicular stomatitis virus nucleocapsid in complex with the nucleocapsid-binding domain of the small polymerase cofactor, P. Proc. Natl. Acad. Sci. USA 2009, 106, 11713–11718. [Google Scholar] [CrossRef] [Green Version]
- Leyrat, C.; Yabukarski, F.; Tarbouriech, N.; Ribeiro, E.A., Jr.; Jensen, M.R.; Blackledge, M.; Ruigrok, R.W.; Jamin, M. Structure of the vesicular stomatitis virus N(0)-P complex. PLoS Pathog. 2011, 7, e1002248. [Google Scholar] [CrossRef] [PubMed]
- Yabukarski, F.; Lawrence, P.; Tarbouriech, N.; Bourhis, J.M.; Delaforge, E.; Jensen, M.R.; Ruigrok, R.W.; Blackledge, M.; Volchkov, V.; Jamin, M. Structure of Nipah virus unassembled nucleoprotein in complex with its viral chaperone. Nat. Struct. Mol. Biol. 2014, 21, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Guryanov, S.G.; Liljeroos, L.; Kasaragod, P.; Kajander, T.; Butcher, S.J. Crystal structure of the measles virus nucleoprotein core in complex with an N-terminal region of phosphoprotein. J. Virol. 2015, 90, 2849–2857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, D.W.; Borek, D.; Luthra, P.; Binning, J.M.; Anantpadma, M.; Liu, G.; Harvey, I.B.; Su, Z.; Endlich-Frazier, A.; Pan, J.; et al. An intrinsically disordered peptide from Ebola virus VP35 controls viral RNA synthesis by modulating nucleoprotein-RNA interactions. Cell Rep. 2015, 11, 376–389. [Google Scholar] [CrossRef] [Green Version]
- Renner, M.; Bertinelli, M.; Leyrat, C.; Paesen, G.C.; Saraiva de Oliveira, L.F.; Huiskonen, J.T.; Grimes, J.M. Nucleocapsid assembly in pneumoviruses is regulated by conformational switching of the N protein. Elife 2016, 5, e12627. [Google Scholar] [CrossRef]
- Zhu, T.; Song, H.; Peng, R.; Shi, Y.; Qi, J.; Gao, G.F. Crystal structure of the Marburg virus nucleoprotein core domain chaperoned by a VP35 peptide reveals a conserved drug target for Filovirus. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Aggarwal, M.; Leser, G.P.; Kors, C.A.; Lamb, R.A. Structure of the paramyxovirus parainfluenza virus 5 nucleoprotein in complex with an amino-terminal peptide of the phosphoprotein. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Wang, X.; Xie, M.; Wu, W.; Chen, Z. Structural basis of human parainfluenza virus 3 unassembled nucleoprotein in complex with its viral chaperone. J. Virol. 2022, 96, e0164821. [Google Scholar] [CrossRef]
- Barik, S.; Banerjee, A.K. Phosphorylation by cellular casein kinase II is essential for transcriptional activity of vesicular stomatitis virus phosphoprotein P. Proc. Natl. Acad. Sci. USA 1992, 89, 6570–6574. [Google Scholar] [CrossRef] [Green Version]
- Schwemmle, M.; De, B.; Shi, L.; Banerjee, A.; Lipkin, W.I. Borna disease virus P-protein is phosphorylated by protein kinase Cepsilon and casein kinase II. J. Biol. Chem. 1997, 272, 21818–21823. [Google Scholar] [CrossRef] [Green Version]
- Dupuy, L.C.; Dobson, S.; Bitko, V.; Barik, S. Casein kinase 2-mediated phosphorylation of respiratory syncytial virus phosphoprotein P is essential for the transcription elongation activity of the viral polymerase; phosphorylation by casein kinase 1 occurs mainly at Ser(215) and is without effect. J. Virol. 1999, 73, 8384–8392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raux, H.; Flamand, A.; Blondel, D. Interaction of the rabies virus P protein with the LC8 dynein light chain. J. Virol. 2000, 74, 10212–10216. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Fuentes, S.M.; Timani, K.; Sun, D.; Murphy, C.; Lin, Y.; August, A.; Teng, M.N.; He, B. Akt plays a critical role in replication of nonsegmented negative-stranded RNA viruses. J. Virol. 2008, 82, 105–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, T.; Matsuoka, M.; Chang, T.H.; Bray, M.; Jones, S.; Tashiro, M.; Kato, A.; Ozato, K. Ebolavirus VP35 interacts with the cytoplasmic dynein light chain 8. J. Virol. 2009, 83, 6952–6956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, D.W.; Ginder, N.D.; Fulton, D.B.; Nix, J.; Basler, C.F.; Honzatko, R.B.; Amarasinghe, G.K. Structure of the Ebola VP35 interferon inhibitory domain. Proc. Natl. Acad. Sci. USA 2009, 106, 411–416. [Google Scholar] [CrossRef] [Green Version]
- Lahaye, X.; Vidy, A.; Fouquet, B.; Blondel, D. Hsp70 protein positively regulates rabies virus infection. J. Virol. 2012, 86, 4743–4751. [Google Scholar] [CrossRef] [Green Version]
- Fouquet, B.; Nikolic, J.; Larrous, F.; Bourhy, H.; Wirblich, C.; Lagaudrière-Gesbert, C.; Blondel, D. Focal adhesion kinase is involved in rabies virus infection through its interaction with viral phosphoprotein P. J. Virol. 2015, 89, 1640–1651. [Google Scholar] [CrossRef] [Green Version]
- Jespersen, N.E.; Leyrat, C.; Gerard, F.C.; Bourhis, J.M.; Blondel, D.; Jamin, M.; Barbar, E. The LC8-RavP ensemble structure svinces a role for LC8 in regulating lyssavirus polymerase functionality. J. Mol. Biol. 2019, 431, 4959–4977. [Google Scholar] [CrossRef]
- Briggs, K.; Wang, L.; Nagashima, K.; Zengel, J.; Tripp, R.A.; He, B. Regulation of mumps virus replication and transcription by kinase RPS6KB1. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Lim, D.; Shin, H.C.; Choi, J.S.; Kim, S.J.; Ku, B. Crystal structure of human LC8 bound to a peptide from Ebola virus VP35. J. Microbiol. 2021, 59, 410–416. [Google Scholar] [CrossRef]
- Kuhn, J.H.; Durrwald, R.; Bao, Y.; Briese, T.; Carbone, K.; Clawson, A.N.; deRisi, J.L.; Garten, W.; Jahrling, P.B.; Kolodziejek, J.; et al. Taxonomic reorganization of the family Bornaviridae. Arch. Virol. 2015, 160, 621–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyndman, T.H.; Shilton, C.M.; Stenglein, M.D.; Wellehan, J.F.X., Jr. Divergent bornaviruses from Australian carpet pythons with neurological disease date the origin of extant Bornaviridae prior to the end-Cretaceous extinction. PLoS Pathog. 2018, 14, e1006881. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.D.; Chen, X.; Tian, J.H.; Chen, L.J.; Li, K.; Wang, W.; Eden, J.S.; Shen, J.J.; Liu, L.; et al. The evolutionary history of vertebrate RNA viruses. Nature 2018, 556, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Hayashi, Y.; Omori, H.; Honda, T.; Daito, T.; Horie, M.; Ikuta, K.; Fujino, K.; Nakamura, S.; Schneider, U.; et al. Bornavirus closely associates and segregates with host chromosomes to ensure persistent intranuclear infection. Cell Host Microbe 2012, 11, 492–503. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, B.; Tappe, D.; Hoper, D.; Herden, C.; Boldt, A.; Mawrin, C.; Niederstrasser, O.; Muller, T.; Jenckel, M.; van der Grinten, E.; et al. A variegated squirrel bornavirus associated with fatal human encephalitis. N. Engl. J. Med. 2015, 373, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Korn, K.; Coras, R.; Bobinger, T.; Herzog, S.M.; Lucking, H.; Stohr, R.; Huttner, H.B.; Hartmann, A.; Ensser, A. Fatal encephalitis associated with Borna disease virus 1. N. Engl. J. Med. 2018, 379, 1375–1377. [Google Scholar] [CrossRef]
- Cadar, D.; Allendorf, V.; Schulze, V.; Ulrich, R.G.; Schlottau, K.; Ebinger, A.; Hoffmann, B.; Hoffmann, D.; Rubbenstroth, D.; Ismer, G.; et al. Introduction and spread of variegated squirrel bornavirus 1 (VSBV-1) between exotic squirrels and spill-over infections to humans in Germany. Emerg. Microbes Infect. 2021, 10, 602–611. [Google Scholar] [CrossRef]
- Hirai, Y.; Tomonaga, K.; Horie, M. Borna disease virus phosphoprotein triggers the organization of viral inclusion bodies by liquid-liquid phase separation. Int. J. Biol. Macromol. 2021, 192, 55–63. [Google Scholar] [CrossRef]
- Marty, F.H.; Bettamin, L.; Thouard, A.; Bourgade, K.; Allart, S.; Larrieu, G.; Malnou, C.E.; Gonzalez-Dunia, D.; Suberbielle, E. Borna disease virus docks on neuronal DNA double-strand breaks to replicate and dampens neuronal activity. iScience 2022, 25, 103621. [Google Scholar] [CrossRef]
- Suberbielle, E.; Stella, A.; Pont, F.; Monnet, C.; Mouton, E.; Lamouroux, L.; Monsarrat, B.; Gonzalez-Dunia, D. Proteomic analysis reveals selective impediment of neuronal remodeling upon Borna disease virus infection. J. Virol. 2008, 82, 12265–12279. [Google Scholar] [CrossRef] [Green Version]
- Bonnaud, E.M.; Szelechowski, M.; Betourne, A.; Foret, C.; Thouard, A.; Gonzalez-Dunia, D.; Malnou, C.E. Borna disease virus phosphoprotein modulates epigenetic signaling in neurons to control viral replication. J. Virol. 2015, 89, 5996–6008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudolph, M.G.; Kraus, I.; Dickmanns, A.; Eickmann, M.; Garten, W.; Ficner, R. Crystal structure of the borna disease virus nucleoprotein. Structure 2003, 11, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Neumann, P.; Lieber, D.; Meyer, S.; Dautel, P.; Kerth, A.; Kraus, I.; Garten, W.; Stubbs, M.T. Crystal structure of the Borna disease virus matrix protein (BDV-M) reveals ssRNA binding properties. Proc. Natl. Acad. Sci. USA 2009, 106, 3710–3715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabsch, W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 1993, 26, 795–800. [Google Scholar] [CrossRef]
- Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 2, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Tickle, I.J.; Flensburg, C.; Keller, P.; Paciorek, W.; Sharff, A.; Vonrhein, C.; Bricogne, G. STARANISO; Global Phasing Ltd.: Cambridge, UK, 2017. [Google Scholar]
- Vonrhein, C.; Flensburg, C.; Keller, P.; Sharff, A.; Smart, O.; Paciorek, W.; Womack, T.; Bricogne, G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr. 2011, 67 Pt 4, 293–302. [Google Scholar] [CrossRef] [Green Version]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40 Pt 4, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Terwilliger, T.C.; Grosse-Kunstleve, R.W.; Afonine, P.V.; Moriarty, N.W.; Zwart, P.H.; Hung, L.W.; Read, R.J.; Adams, P.D. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr. 2008, 64 Pt 1, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 2, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 4, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.X.; Ben Jelloul, M.; Long, F.; Vagin, A.; Knipscheer, P.; Lebbink, J.; Sixma, T.K.; Lamzin, V.S.; Murshudov, G.N.; Perrakis, A. ARP/wARP and molecular replacement: The next generation. Acta Crystallogr. D Biol. Crystallogr. 2008, 64 Pt 1, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Langer, G.; Cohen, S.X.; Lamzin, V.S.; Perrakis, A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 2008, 3, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997, 53 Pt 3, 240–255. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64 Pt 1, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Cowtan, K. Recent developments in classical density modification. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 4, 470–478. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Schrodinger, LLC. The PyMOL Molecular Graphics System, Version 1.8; Schrödinger, Inc.: New York, NY, USA, 2015.
- Thureau, A.; Roblin, P.; Perez, J. BioSAXS on the SWING beamline at Synchrotron SOLEIL. J. Appl. Crystallogr. 2021, 54, 1698–1710. [Google Scholar] [CrossRef]
- Manalastas-Cantos, K.; Konarev, P.V.; Hajizadeh, N.R.; Kikhney, A.G.; Petoukhov, M.V.; Molodenskiy, D.S.; Panjkovich, A.; Mertens, H.D.T.; Gruzinov, A.; Borges, C.; et al. ATSAS 3.0: Expanded functionality and new tools for small-angle scattering data analysis. J. Appl. Crystallogr. 2021, 54, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, J.B.; Gillilan, R.E.; Skou, S. BioXTAS RAW: Improvements to a free open-source program for small-angle X-ray scattering data reduction and analysis. J. Appl. Crystallogr. 2017, 50, 1545–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svergun, D. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 1992, 25, 495–503. [Google Scholar] [CrossRef]
- Hansen, S. Bayesian estimation of hyperparameters for indirect Fourier transformation in small-angle scattering. J. Appl. Crystallogr. 2000, 33, 1415–1421. [Google Scholar] [CrossRef]
- Rambo, R.P.; Tainer, J.A. Accurate assessment of mass, models and resolution by small-angle scattering. Nature 2013, 496, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Franke, D.; Svergun, D.I. DAMMIF, a program for rapid ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2009, 42 Pt 2, 342–346. [Google Scholar] [CrossRef] [Green Version]
- Volkov, V.V.; Svergun, D.I. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2003, 36, 860–864. [Google Scholar] [CrossRef] [Green Version]
- Svergun, D.I. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 1999, 76, 2879–2886. [Google Scholar] [CrossRef] [Green Version]
- Tria, G.; Mertens, H.D.; Kachala, M.; Svergun, D.I. Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ 2015, 2 Pt 2, 207–217. [Google Scholar] [CrossRef] [Green Version]
- Moore, B.L.; Kelley, L.A.; Barber, J.; Murray, J.W.; MacDonald, J.T. High-quality protein backbone reconstruction from alpha carbons using Gaussian mixture models. J. Comput. Chem. 2013, 34, 1881–1889. [Google Scholar] [CrossRef]
- Krivov, G.G.; Shapovalov, M.V.; Dunbrack, R.L., Jr. Improved prediction of protein side-chain conformations with SCWRL4. Proteins 2009, 77, 778–795. [Google Scholar] [CrossRef] [Green Version]
- Kanda, T.; Horie, M.; Komatsu, Y.; Tomonaga, K. The Borna disease virus 2 (BoDV-2) nucleoprotein is a conspecific protein that enhances BoDV-1 RNA-dependent RNA polymerase activity. J. Virol. 2021, 95, e0093621. [Google Scholar] [CrossRef] [PubMed]
- Reuter, A.; Horie, M.; Höper, D.; Ohnemus, A.; Narr, A.; Rinder, M.; Beer, M.; Staeheli, P.; Rubbenstroth, D. Synergistic antiviral activity of ribavirin and interferon-α against parrot bornaviruses in avian cells. J. Gen. Virol. 2016, 97, 2096–2103. [Google Scholar] [CrossRef]
- Tu, Y.X.I.; Sydor, A.M.; Coyaud, E.; Laurent, E.M.N.; Dyer, D.; Mellouk, N.; St-Germain, J.; Vernon, R.M.; Forman-Kay, J.D.; Li, T.; et al. Global proximity interactome of the human macroautophagy pathway. Autophagy 2022, 18, 1174–1186. [Google Scholar] [CrossRef] [PubMed]
- Gerard, F.C.; Ribeiro Ede, A., Jr.; Leyrat, C.; Ivanov, I.; Blondel, D.; Longhi, S.; Ruigrok, R.W.; Jamin, M. Modular organization of rabies virus phosphoprotein. J. Mol. Biol. 2009, 388, 978–996. [Google Scholar] [CrossRef] [PubMed]
- Crick, F.H.C. The Fourier transform of a coiled-coil. Acta Crystallogr. 1953, 6, 685–689. [Google Scholar] [CrossRef] [Green Version]
- Crick, F.H.C. The packing of α-helices: Simple coiled-coils. Acta Crystallogr. 1953, 6, 689–697. [Google Scholar] [CrossRef] [Green Version]
- Grigoryan, G.; Degrado, W.F. Probing designability via a generalized model of helical bundle geometry. J. Mol. Biol. 2011, 405, 1079–1100. [Google Scholar] [CrossRef] [Green Version]
- Strelkov, S.V.; Burkhard, P. Analysis of alpha-helical coiled-coils with the program TWISTER reveals a structural mechanism for stutter compensation. J. Struct. Biol. 2002, 137, 54–64. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, M.D.; Mendler, C.T.; Bassler, J.; Karamichali, I.; Ridderbusch, O.; Lupas, A.N.; Hernandez Alvarez, B. alpha/beta coiled-coils. Elife 2016, 5, 351–358. [Google Scholar] [CrossRef]
- Kliche, S.; Stitz, L.; Mangalam, H.; Shi, L.; Binz, T.; Niemann, H.; Briese, T.; Lipkin, W.I. Characterization of the Borna disease virus phosphoprotein, p23. J. Virol. 1996, 70, 8133–8137. [Google Scholar] [CrossRef] [Green Version]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.I.; Birendra, K.C.; Zhu, W.; Motamedchaboki, K.; Doye, V.; Roux, K.J. Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc. Natl. Acad. Sci. USA 2014, 111, E2453–E2461. [Google Scholar] [CrossRef] [Green Version]
- Mehus, A.A.; Anderson, R.H.; Roux, K.J. BioID Identification of Lamin-Associated Proteins. Methods Enzymol. 2016, 569, 3–22. [Google Scholar]
- Kamitani, W.; Shoya, Y.; Kobayashi, T.; Watanabe, M.; Lee, B.J.; Zhang, G.; Tomonaga, K.; Ikuta, K. Borna disease virus phosphoprotein binds a neurite outgrowth factor, amphoterin/HMG-1. J. Virol. 2001, 75, 8742–8751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Kobayashi, T.; Kamitani, W.; Komoto, S.; Yamashita, M.; Baba, S.; Yanai, H.; Ikuta, K.; Tomonaga, K. Borna disease virus phosphoprotein represses p53-mediated transcriptional activity by interference with HMGB1. J. Virol. 2003, 77, 12243–12251. [Google Scholar] [CrossRef] [PubMed]
- Starokadomskyy, P.; Gluck, N.; Li, H.; Chen, B.; Wallis, M.; Maine, G.N.; Mao, X.; Zaidi, I.W.; Hein, M.Y.; McDonald, F.J.; et al. CCDC22 deficiency in humans blunts activation of proinflammatory NF-kappaB signaling. J. Clin. Investig. 2013, 123, 2244–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoh, S.M.; Schneider, M.; Seifried, J.; Soonthornvacharin, S.; Akleh, R.E.; Olivieri, K.C.; De Jesus, P.D.; Ruan, C.; de Castro, E.; Ruiz, P.A.; et al. PQBP1 Is a proximal sensor of the cGAS-dependent innate response to HIV-1. Cell 2015, 161, 1293–1305. [Google Scholar] [CrossRef] [Green Version]
- Atzei, P.; Gargan, S.; Curran, N.; Moynagh, P.N. Cactin targets the MHC class III protein IkappaB-like (IkappaBL) and inhibits NF-kappaB and interferon-regulatory factor signaling pathways. J. Biol. Chem. 2010, 285, 36804–36817. [Google Scholar] [CrossRef] [Green Version]
- Morchikh, M.; Cribier, A.; Raffel, R.; Amraoui, S.; Cau, J.; Severac, D.; Dubois, E.; Schwartz, O.; Bennasser, Y.; Benkirane, M. HEXIM1 and NEAT1 long non-coding RNA form a multi-subunit complex that regulates DNA-mediated innate immune response. Mol. Cell 2017, 67, 387–399.e5. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Zhang, Q.; Zhang, R.; Lu, Y.; Wang, X.; Tian, H.; Yang, Y.; Gu, Z.; Gao, Y.; Yang, X.; et al. N(6)-methyladenosine RNA modification suppresses antiviral innate sensing pathways via reshaping double-stranded RNA. Nat. Commun. 2021, 12, 1582. [Google Scholar] [CrossRef] [PubMed]
- Schneider, U.; Blechschmidt, K.; Schwemmle, M.; Staeheli, P. Overlap of interaction domains indicates a central role of the P protein in assembly and regulation of the Borna disease virus polymerase complex. J. Biol. Chem. 2004, 279, 55290–55296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilman, M.S.A.; Furmanova-Hollenstein, P.; Pascual, G.; B van’t Wout, A.; Langedijk, J.P.M.; McLellan, J.S. Transient opening of trimeric prefusion RSV F proteins. Nat. Commun. 2019, 10, 2105. [Google Scholar] [CrossRef] [Green Version]
- Cortez, D. Replication-Coupled DNA Repair. Mol. Cell 2019, 74, 866–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qing, X.; Zhang, G.; Wang, Z.Q. DNA damage response in neurodevelopment and neuromaintenance. FEBS J. 2022. [Google Scholar] [CrossRef] [PubMed]
- Suberbielle, E.; Sanchez, P.E.; Kravitz, A.V.; Wang, X.; Ho, K.; Eilertson, K.; Devidze, N.; Kreitzer, A.C.; Mucke, L. Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-beta. Nat. Neurosci. 2013, 16, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Suberbielle, E.; Djukic, B.; Evans, M.; Kim, D.H.; Taneja, P.; Wang, X.; Finucane, M.; Knox, J.; Ho, K.; Devidze, N.; et al. DNA repair factor BRCA1 depletion occurs in Alzheimer brains and impairs cognitive function in mice. Nat. Commun. 2015, 6, 8897. [Google Scholar] [CrossRef] [Green Version]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Garcia-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]
- Goedhart, J.; Luijsterburg, M.S. VolcaNoseR is a web app for creating, exploring, labeling and sharing volcano plots. Sci Rep. 2020, 10, 20560. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Gouet, P.; Courcelle, E.; Stuart, D.I.; Metoz, F. ESPript: Analysis of multiple sequence alignments in PostScript. Bioinformatics 1999, 15, 305–308. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarbouriech, N.; Chenavier, F.; Kawasaki, J.; Bachiri, K.; Bourhis, J.-M.; Legrand, P.; Freslon, L.L.; Laurent, E.M.N.; Suberbielle, E.; Ruigrok, R.W.H.; et al. Borna Disease Virus 1 Phosphoprotein Forms a Tetramer and Interacts with Host Factors Involved in DNA Double-Strand Break Repair and mRNA Processing. Viruses 2022, 14, 2358. https://doi.org/10.3390/v14112358
Tarbouriech N, Chenavier F, Kawasaki J, Bachiri K, Bourhis J-M, Legrand P, Freslon LL, Laurent EMN, Suberbielle E, Ruigrok RWH, et al. Borna Disease Virus 1 Phosphoprotein Forms a Tetramer and Interacts with Host Factors Involved in DNA Double-Strand Break Repair and mRNA Processing. Viruses. 2022; 14(11):2358. https://doi.org/10.3390/v14112358
Chicago/Turabian StyleTarbouriech, Nicolas, Florian Chenavier, Junna Kawasaki, Kamel Bachiri, Jean-Marie Bourhis, Pierre Legrand, Lily L. Freslon, Estelle M. N. Laurent, Elsa Suberbielle, Rob W. H. Ruigrok, and et al. 2022. "Borna Disease Virus 1 Phosphoprotein Forms a Tetramer and Interacts with Host Factors Involved in DNA Double-Strand Break Repair and mRNA Processing" Viruses 14, no. 11: 2358. https://doi.org/10.3390/v14112358