
In Silico Genome Analysis Reveals the Evolution and Potential Impact of SARS-CoV-2 Omicron Structural Changes on Host Immune Evasion and Antiviral Therapeutics
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. SARS-CoV-2 Data Analyses
2.2. SARS-CoV-2 Target Protein and Drug Interaction Analysis
2.3. Prediction of T-Cell Epitopes (TCEs) and Their Binding Affinity to Predominant HLA Alleles
3. Results and Discussion
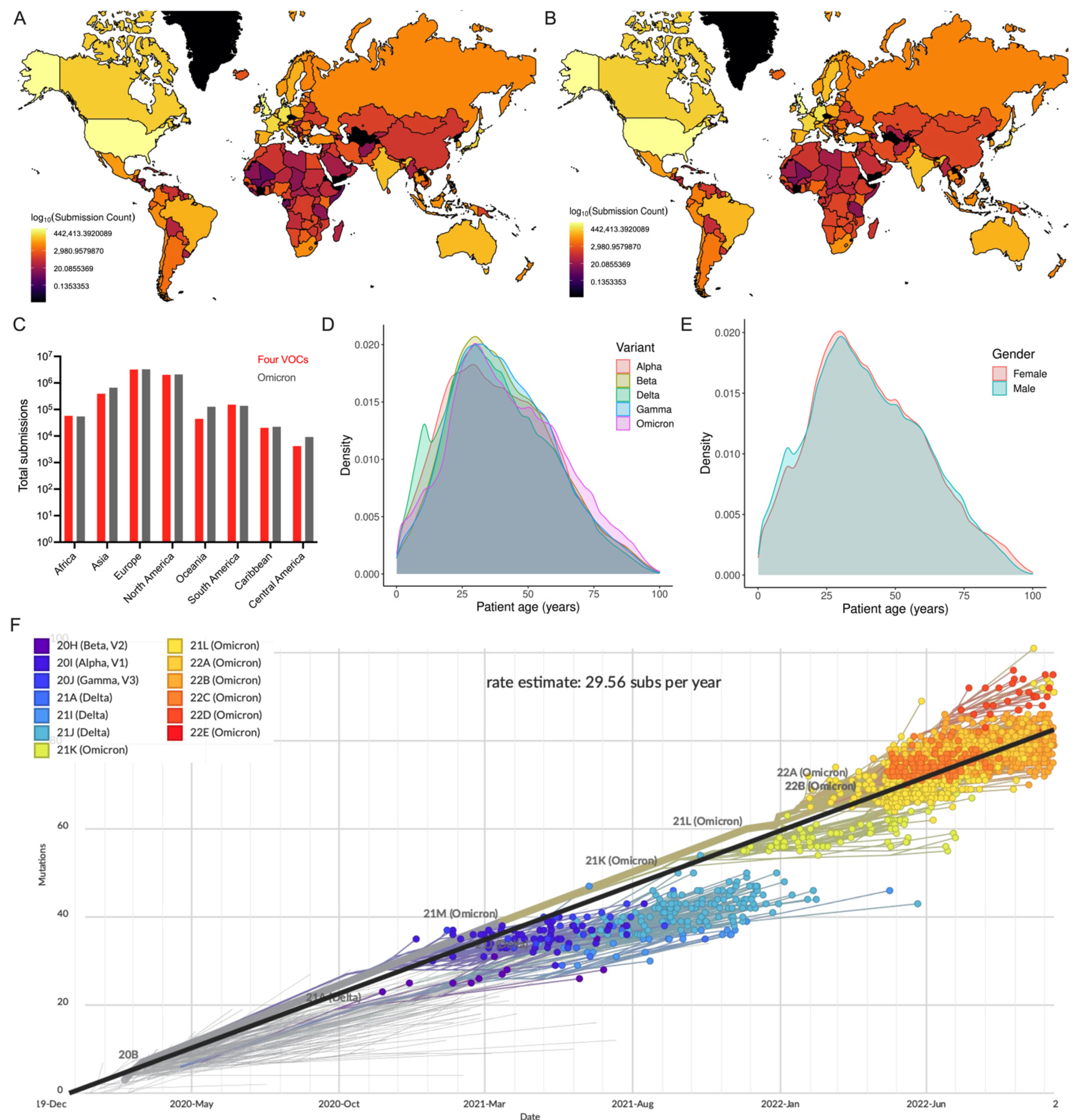
3.1. Geographical Distribution of Omicron Variant
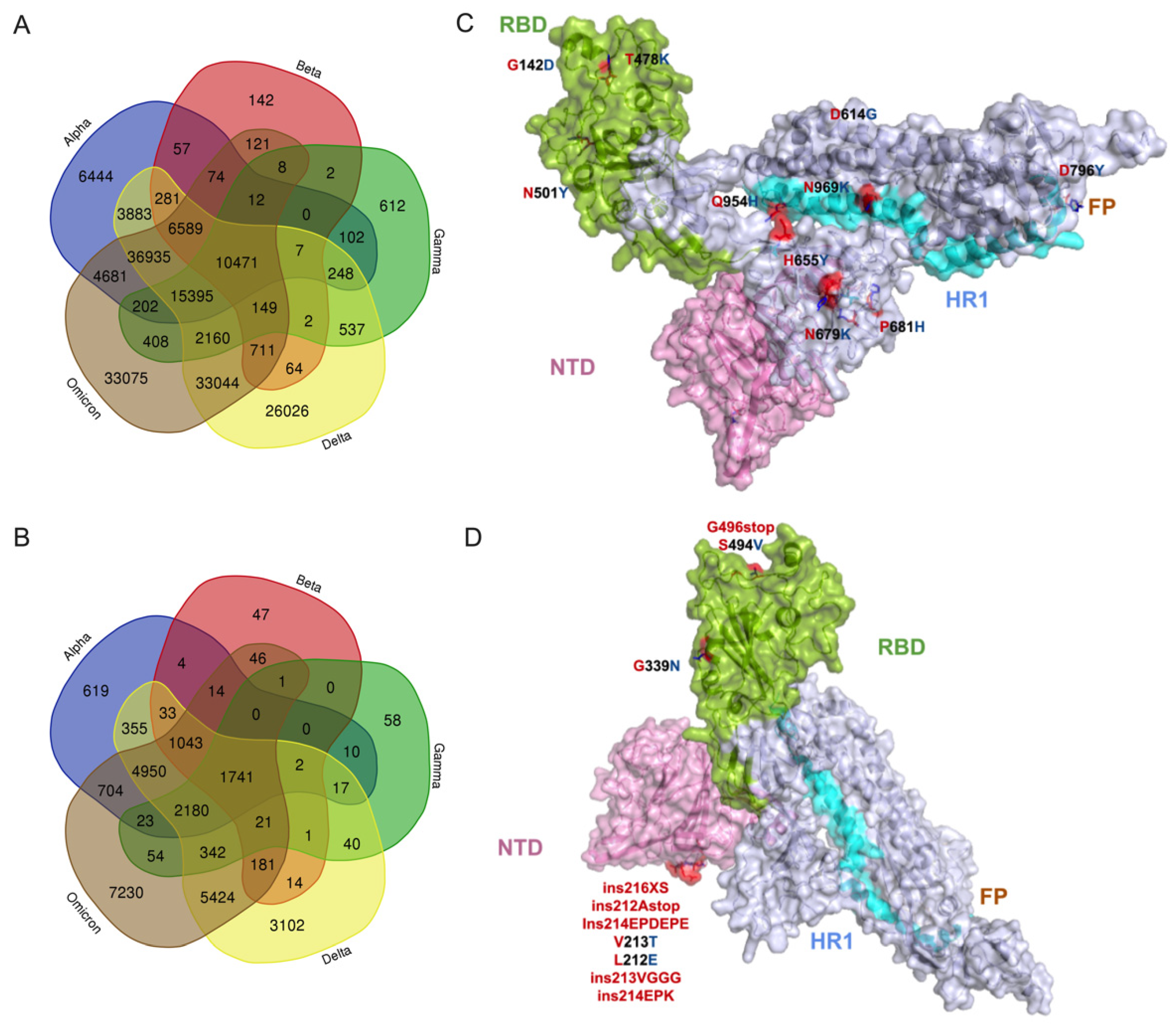
3.2. Potential Effect of Omicron Mutations on Host Immune Response
3.3. Molecular Docking Analysis of Omicron Proteins with Known Antiviral Drugs
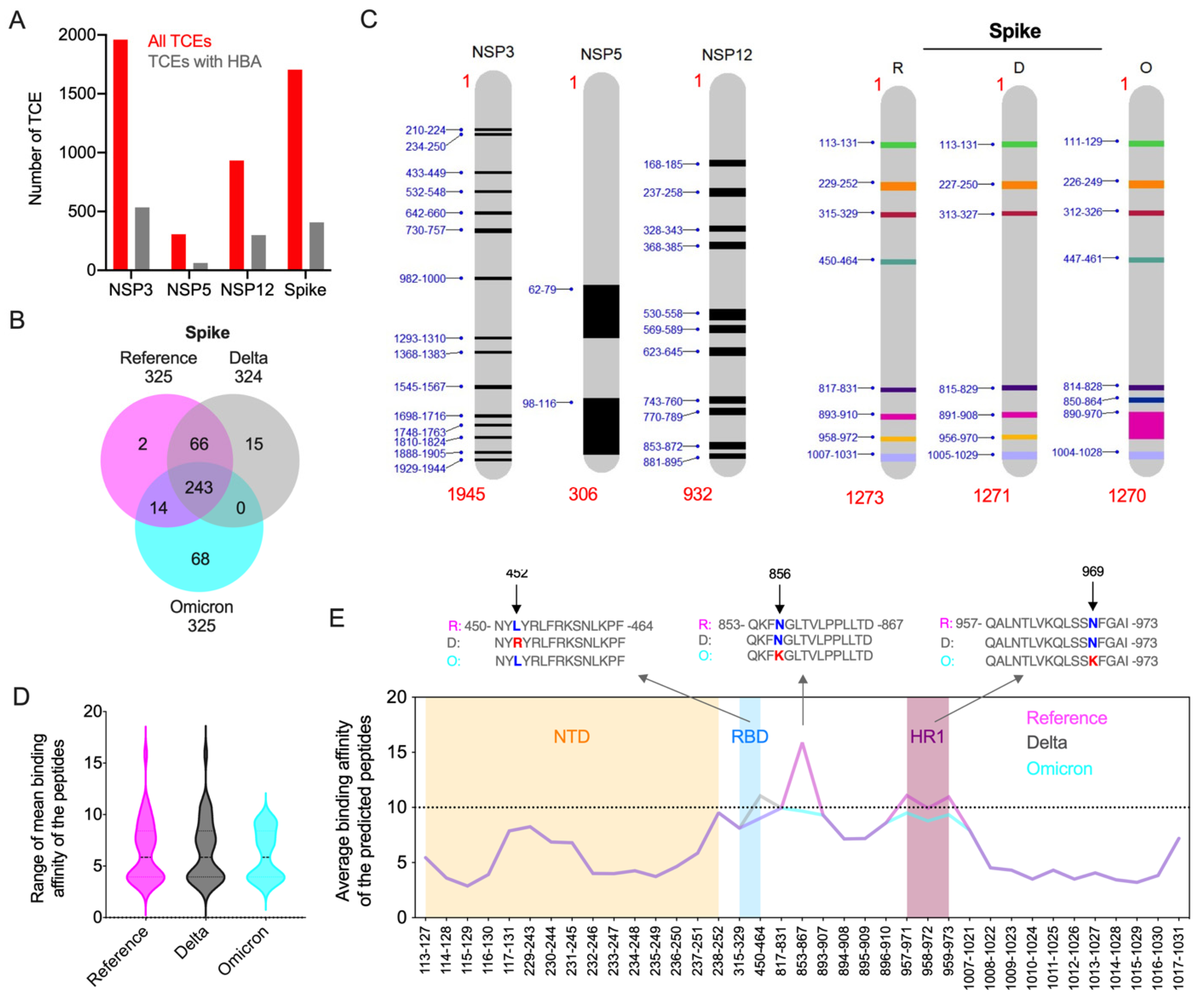
3.4. Impact of Key Amino Acid and Structural Changes in T-Cell Epitopes in Modulating the Host Cellular Immune Response
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical Features of Patients Infected with 2019 Novel Coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Ramaiah, A.; Arumugaswami, V. Insights into Cross-Species Evolution of Novel Human Coronavirus SARS-CoV-2 and Defining Immune Determinants for Vaccine Development. bioRxiv 2020. 2020.01.29.925867. [Google Scholar] [CrossRef] [Green Version]
- Kannan, S.R.; Spratt, A.N.; Sharma, K.; Chand, H.S.; Byrareddy, S.N.; Singh, K. Omicron SARS-CoV-2 Variant: Unique Features and Their Impact on Pre-Existing Antibodies. J. Autoimmun. 2022, 126, 102779. [Google Scholar] [CrossRef] [PubMed]
- WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/?adgroupsurvey={adgroupsurvey}&gclid=Cj0KCQjw7KqZBhCBARIsAI-fTKJFN2-Suk6nKcfohSYp0SPeXye_qAQ5Wv-ou27miS3x3RhSkhh9SnMaAlTpEALw_wcB (accessed on 25 August 2022).
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9, 1542. [Google Scholar] [CrossRef] [PubMed]
- Edara, V.-V.; Pinsky, B.A.; Suthar, M.S.; Lai, L.; Davis-Garner, M.E.; Floyd, K.; Flowers, M.W.; Wrammert, J.; Hussaini, L.; Ciric, C.R.; et al. Infection and Vaccine-Induced Neutralizing Antibody Responses to the SARS-CoV-2 B.1.617.1 Variants. N. Engl. J. Med. 2021, 385, 664–666. [Google Scholar] [CrossRef]
- Hoffmann, M.; Hofmann-Winkler, H.; Krüger, N.; Kempf, A.; Nehlmeier, I.; Graichen, L.; Arora, P.; Sidarovich, A.; Moldenhauer, A.-S.; Winkler, M.S.; et al. SARS-CoV-2 Variant B.1.617 Is Resistant to Bamlanivimab and Evades Antibodies Induced by Infection and Vaccination. Cell Rep. 2021, 36, 109415. [Google Scholar] [CrossRef]
- Leung, K.; Shum, M.H.; Leung, G.M.; Lam, T.T.; Wu, J.T. Early Transmissibility Assessment of the N501Y Mutant Strains of SARS-CoV-2 in the United Kingdom, October to November 2020. Eurosurveillance 2021, 26, 2002106. [Google Scholar] [CrossRef]
- Mlcochova, P.; Kemp, S.A.; Dhar, M.S.; Papa, G.; Meng, B.; Ferreira, I.A.T.M.; Datir, R.; Collier, D.A.; Albecka, A.; Singh, S.; et al. SARS-CoV-2 B.1.617.2 Delta Variant Replication and Immune Evasion. Nature 2021, 599, 114–119. [Google Scholar] [CrossRef]
- Yadav, P.D.; Sapkal, G.N.; Abraham, P.; Ella, R.; Deshpande, G.; Patil, D.Y.; Nyayanit, D.A.; Gupta, N.; Sahay, R.R.; Shete, A.M.; et al. Neutralization of Variant under Investigation B.1.617 with Sera of BBV152 Vaccinees. Clin. Infect. Dis. 2021, 74, 366–368. [Google Scholar] [CrossRef]
- Faria, N.R.; Claro, I.M.; Candido, D.; Franco, L.A.M.; Andrade, P.S.; Coletti, T.M.; Silva, C.A.M.; Sales, F.C.; Manuli, E.R.; Aguiar, R.S.; et al. Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in Manaus: Preliminary Findings. Available online: https://virological.org/t/genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-manaus-preliminary-findings/586 (accessed on 5 January 2022).
- CDC. COVID-19. Available online: https://public4.pagefreezer.com/browse/CDC%20Covid%20Pages/15-07-2022T12:20/https://www.cdc.gov/coronavirus/2019-ncov/science/science-briefs/scientific-brief-omicron-variant.html (accessed on 20 December 2021).
- Espenhain, L.; Funk, T.; Overvad, M.; Edslev, S.M.; Fonager, J.; Ingham, A.C.; Rasmussen, M.; Madsen, S.L.; Espersen, C.H.; Sieber, R.N.; et al. Epidemiological Characterisation of the First 785 SARS-CoV-2 Omicron Variant Cases in Denmark, December 2021. Eurosurveillance 2021, 26, 2101146. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cheng, G. Sequence Analysis of the Emerging SARS-CoV-2 Variant Omicron in South Africa. J. Med. Virol. 2021, 94, 1728–1733. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamalipour Soufi, G.; Iravani, S. Potential Inhibitors of SARS-CoV-2: Recent Advances. J. Drug Target. 2020, 29, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Mengist, H.M.; Dilnessa, T.; Jin, T. Structural Basis of Potential Inhibitors Targeting SARS-CoV-2 Main Protease. Front. Chem. 2021, 9, 622898. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Marak, B.N.; Dowarah, J.; Khiangte, L.; Singh, V.P. Step toward Repurposing Drug Discovery for COVID-19 Therapeutics through in Silico Approach. Drug Dev. Res. 2020, 82, 374–392. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand—Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Jensen, K.K.; Andreatta, M.; Marcatili, P.; Buus, S.; Greenbaum, J.A.; Yan, Z.; Sette, A.; Peters, B.; Nielsen, M. Improved Methods for Predicting Peptide Binding Affinity to MHC Class II Molecules. Immunology 2018, 154, 394–406. [Google Scholar] [CrossRef]
- Ramaiah, A.; Koralur, M.C.; Dasch, G.A. Complexity of Type-Specific 56 KDa Antigen CD4 T-Cell Epitopes of Orientia Tsutsugamushi Strains Causing Scrub Typhus in India. PLoS ONE 2018, 13, e0196240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, M.I.; MacGowan, S.A.; Kutuzov, M.A.; Dushek, O.; Barton, G.J.; van der Merwe, P.A. Effects of Common Mutations in the SARS-CoV-2 Spike RBD and Its Ligand, the Human ACE2 Receptor on Binding Affinity and Kinetics. eLife 2021, 10, e70658. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.-J.; Guo, H.; Luo, G. Omicron variant (B.1.1.529) of SARS-CoV-2, a global urgent public health altert! J. Med. Virol. 2022, 94, 1255–1256. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; He, P.; Li, J.; Liu, H.; Shi, M.; Yu, J.; Wei, H. Steep decline in binding capability of SARS-CoV-2 omicron variant (B.1.1.529) RBD to the antibodies in early COVID-19 convalescent sera and inactivated vaccine sera. Viruses 2022, 14, 335. [Google Scholar] [CrossRef]
- Chakravarty, N.; Senthilnathan, T.; Paiola, S.; Gyani, P.; Cario, C.; Urena, E.; Jeysankar, A.; Jeysankar, P.; Ignatius Irudayam, J.; Subramanian, N.; et al. Neurological Pathophysiology of SARSCoV2 and Pandemic Potential RNA Viruses: A Comparative Analysis. FEBS Lett. 2021, 595, 2854–2871. [Google Scholar] [CrossRef]
- Mariano, G.; Farthing, R.J.; Lale-Farjat, S.L.; Bergeron, J.R. Bergeron, Structural Characterization of SARSCoV2: Where We Are, and Where We Need to Be. Front. Mol. Biosci. 2020, 7, 605236. [Google Scholar] [CrossRef]
- Pachetti, M.; Marini, B.; Benedetti, F.; Giudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.C.; et al. Emerging SARSCoV2 Mutation Hot Spots Include a Novel RNA-dependent-RNA Polymerase Variant. J. Transl. Med. 2020, 18, 179. [Google Scholar] [CrossRef] [Green Version]
- Ullrich, S.; Nitsche, C. The SARSCoV2 Main Protease as Drug Target. Bioorganic Med. Chem. Lett. 2020, 30, 127377. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Drug | Variant | Binding Affinity (kcal/mol) | Residues Involved in Interactions |
|---|---|---|---|---|
| nsp3 | Parecoxib | Reference | −8.4 | Ala242, Gly250, Gly251, Gly252, Val253, Ala333, Gly334, Ile335, Phe336, Ala358, Phe360, Leu364, Val539 |
| Delta | −8.4 | Val359 | ||
| Omicron | −8.2 | Asp226, Ile227, Ala256, Pro329, Leu330, Val359, Asp361 | ||
| Chlortetracycline | Reference | −8.5 | Asp226, Ile227, Val228, Val253, Ala256, Pro329, Leu330, Ala333, Gly334, Val359, Phe360, Asp361, Leu364 | |
| Delta | −8.5 | No unique residues identified | ||
| Omicron | −8.5 | No unique residues identified | ||
| nsp5 | Nirmatrelvir | Reference | −7.7 | Phe140, Leu141, Asn142, Cys145, His163, Met165, Glu166, His172, Arg188, Gln189, Thr190, Gln192 |
| Delta | −7.6 | His41, Met49, Leu167, Pro168, Asp187 | ||
| Omicron | −7.8 | His41, Met49, Asp187 | ||
| Ivermectin B1b | Reference | −10.2 | Thr26, His41, Ser46, Met49, Asn119, Gly143, Cys145, Met165, Glu166, Pro168, Arg188, Gln189, Thr190, Gln192 | |
| Delta | −9.8 | Thr24, Thr25, Asn142, Ala191 | ||
| Omicron | −10.2 | No unique residues identified | ||
| nsp12 | Sulfasalazine | Reference | −9.6 | Arg583, Gly584, Gly597, Ser592, His599, Asn600, Met601, Lys603, Thr604 |
| Delta | −9.6 | No unique residues identified | ||
| Omicron | −9.3 | No unique residues identified | ||
| Remdesivir | Reference | −6.4 | Arg553, Asp618, Tyr619, Pro620, Lys621, Cys622, Asp623, Asn691, Ser759, Asp760, Asp761, Glu811, Ser814 | |
| Delta | −7.4 | Tyr455, Arg624, Ser682, Thr687 | ||
| Omicron | −8 | Tyr455, Arg533, Lys545, Arg555, Thr556, Trp617, Arg624, Ser682 | ||
| Spike | Ivermectin B1a | Reference | −10 | Lys811, Pro812, Ser813, Arg815, Asp820, Phe823, Gly832, Phe833, Ile834, Lys854, Val860, Thr866, Glu868 |
| Delta | −10 | No unique residues identified | ||
| Omicron | −10 | No unique residues identified | ||
| Atovaquone | Reference | −7.3 | Thr732, Leu828, Ala831, Gly832, Phe833, Ile834, Val860, Pro862 | |
| Delta | −7.3 | No unique residues identified | ||
| Omicron | −7.3 | No unique residues identified |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chauhan, D.; Chakravarty, N.; Jeyachandran, A.V.; Jayakarunakaran, A.; Sinha, S.; Mishra, R.; Arumugaswami, V.; Ramaiah, A. In Silico Genome Analysis Reveals the Evolution and Potential Impact of SARS-CoV-2 Omicron Structural Changes on Host Immune Evasion and Antiviral Therapeutics. Viruses 2022, 14, 2461. https://doi.org/10.3390/v14112461
Chauhan D, Chakravarty N, Jeyachandran AV, Jayakarunakaran A, Sinha S, Mishra R, Arumugaswami V, Ramaiah A. In Silico Genome Analysis Reveals the Evolution and Potential Impact of SARS-CoV-2 Omicron Structural Changes on Host Immune Evasion and Antiviral Therapeutics. Viruses. 2022; 14(11):2461. https://doi.org/10.3390/v14112461
Chicago/Turabian StyleChauhan, Dhruv, Nikhil Chakravarty, Arjit Vijey Jeyachandran, Akshaya Jayakarunakaran, Sanjeev Sinha, Rakesh Mishra, Vaithilingaraja Arumugaswami, and Arunachalam Ramaiah. 2022. "In Silico Genome Analysis Reveals the Evolution and Potential Impact of SARS-CoV-2 Omicron Structural Changes on Host Immune Evasion and Antiviral Therapeutics" Viruses 14, no. 11: 2461. https://doi.org/10.3390/v14112461
APA StyleChauhan, D., Chakravarty, N., Jeyachandran, A. V., Jayakarunakaran, A., Sinha, S., Mishra, R., Arumugaswami, V., & Ramaiah, A. (2022). In Silico Genome Analysis Reveals the Evolution and Potential Impact of SARS-CoV-2 Omicron Structural Changes on Host Immune Evasion and Antiviral Therapeutics. Viruses, 14(11), 2461. https://doi.org/10.3390/v14112461