1. Introduction
Arthropod borne viruses (arboviruses) are characterized by their ability to replicate in both vertebrate and invertebrate hosts. These viruses use the same, limited set of genes to replicate and transmit between the vertebrate host and mosquito vector. Arboviruses are adept at interacting with two distinct hosts, programming efficient replication in both host environments, and priming the progeny for spread to a distinct host environment. There is currently a limited understanding of the molecular determinants of transmission that facilitate efficient alternate infection of host species.
Alphaviruses are arboviruses and are predominantly vectored between vertebrates by mosquitoes. Alphaviruses are divided into two main groups based on the disease they cause. The first group is the arthritogenic viruses, such as Sindbis virus (SINV) and Chikungunya virus (CHIKV), which cause debilitating but non-life-threatening polyarthritis. The second group is the encephalitogenic viruses, such as Venezuelan and Eastern equine encephalitis virus, VEEV/EEEV, which can cause fatal encephalitis. There are currently no effective vaccines for alphaviruses, and outbreaks—particularly of CHIKV—occur regularly around the world. It is therefore important to improve our understanding of alphavirus replication and interaction with both vertebrate and insect hosts. In the work described here, we utilize SINV, the type species of the Alphavirus genus. SINV is an enveloped, single-stranded, positive-sense RNA virus with an approximately 11.7 kb genome. In vertebrate cells, SINV causes an acute, cytolytic infection, whereas in arthropod cells the infection is persistent and noncytolytic.
There is good evidence of alphaviruses’ capabilities to prime the progeny produced in arthropod cells for infection in vertebrate cells, as it has been shown that arthropod-derived alphaviruses have increased infectivity on vertebrate cells, and vertebrate-derived viruses initiate infection of mosquito midgut more efficiently [
1,
2]. Similarly, it has been shown that alphaviruses acquire host-specific alterations that improve their ability to establish infection in the new host; these host-specific differences range from differential packaging of host components such as 18S rRNAs that improve the viral infectivity on mammalian cells to glycosylation of the envelope glycoproteins, which allows for targeted infection of certain mammalian cell types [
2,
3,
4,
5,
6]. Moreover, though a newer field of study, there is evidence of specific RNA modification of viral RNA in the host having various effects on viral RNA function. RNA methylation is driven by host methyltransferase genes, such as METTL3 or TRDMT1. Methyltransferases transfer a methyl group from a methyl carrier molecule, such as S-adenosyl methionine, to certain nucleotides. These methylated nucleotides are recognized by methyl readers that confer different effects on the RNA. Indeed, RNA methylation has been documented to modulate a range of steps in RNA metabolism, including—but not limited to—mRNA stability, translation, localization, and for viral RNA, viral replication [
7,
8,
9,
10,
11,
12,
13,
14,
15,
16,
17]. Recent studies have focused heavily on the role of m6A modifications of viral RNA and how those modifications affect viral replication [
18,
19]. For instance, it has been shown that N6-methyladenosine (m6A) post-transcriptionally regulates the RNA function of the flavivirus hepatitis C virus (HCV) in a negative manner in mammalian cells [
14]. There is also evidence of the hepatitis B virus’ (HBV) life cycle being regulated by m6A modification of the pregenomic RNA intermediate wherein the m6A modifications are required for efficient reverse transcription as well as stabilizing HBV transcripts and ultimately affecting protein expression [
13,
20]. In previous work we showed that SINV acquires 5-methylcytosince (m5C) in mosquito cells [
15]. This m5C modification of SINV RNA was shown to have a proviral effect on the progeny virus used to infect mammalian cells, as ectopic expression of the m5C writer in mosquito cells resulted in a higher specific infectivity as compared to the control [
15]. Further indirect evidence of utilization of host methyltransferases by viruses is provided in multiple reports, showing that virus infection leads to increased expression of methyltransferase genes [
9,
15]. Thus, it is important to explore the role that RNA modification may play in alphavirus infectivity and whether the modification profiles of viral RNA differ between viruses derived from arthropod and mammalian hosts.
Here, we show that in human (HEK-293) cells, mosquito-derived SINV replicates faster and produces more infectious viruses. Mosquito-derived SINV also has increased translation compared to mammalian-derived SINV, and this increased translation is not a result of either incoming genomic RNA stability or increased vRNA levels in the cell. We also show that purified genomic vRNA phenocopies infectivity of purified viruses, and we attribute these differences to host-dependent changes in modification of packaged genomic RNA as determined by LC/MS-MS. Our data support the hypothesis that among other factors [
2,
3,
4], the host-dependent modification profile of the packaged vRNA plays an important role in the efficiency of SINV infection and replication in mammalian cells.
2. Materials and Methods
2.1. Insect and Mammalian Cell Culture
C7/10 Aedes albopictus cells were grown at 28 °C under 5% ambient CO2 in 1× Minimal Essential Media (Corning) supplemented with 10% heat-inactivated fetal bovine serum (Corning), 1% L-glutamine (Corning), 1% non-essential amino acids (Corning), and 1% antibiotic–antimycotic solution (Corning). Vertebrate baby hamster kidney fibroblast or BHK-21 cells, as well as human embryonic kidney epithelial or HEK293 cells, were grown at 37 °C under 5% ambient CO2 in the same 1× Minimal Essential Media (Corning) as the C7/10 cells.
2.2. Obtaining Viruses
BHK-21-derived viruses were obtained by in vitro transcription of SINV full-length plasmid using SP6 RNA polymerase (NEB). IVTs were then transfected into confluent BHK cells using LTX transfection mix (Invitrogen), using manufacturer’s protocol, in a T25 flask (CellStar) with serum-free virus production media (Gibco). Four hours post-transfection, the SF-VPM was removed and replaced with fresh 1× MEM supplemented with 10% FBS. Twenty-four hours post-transfection, the viral supernatant was harvested and purified by centrifugation at 43,000× g for 2.5 h over a 27% w/v sucrose cushion in HNE buffer (150 mM NaCL, 20 mM HEPES, 0.1 mM EDTA). The media was discarded, and the viral pellet was resuspended in 500 µL of HNE buffer. Plaque assays on BHK-21 cells were then performed to determine viral titer.
C7/10-derived virus was obtained by infecting confluent C7/10 cells in a 150 mm dish (CellStar) with P0 BHK-derived virus at an MOI of 0.1 PFU/cell. The inoculum was left on for 3 h, after which the media were replaced, cells were washed with PBS, and fresh 1× MEM was added. Forty-eight hours postinfection, the viral supernatant was harvested, purified and plaque-assayed as previously described.
2.3. Live-Cell Imaging and Single-Step Growth Curve
Live-cell imaging was performed using IncuCyte live-cell analyses system (Essen Biosciences, Ann Arbor, MI, USA). HEK293 cells were plated in a 24-well plate (CellStar) at 70–80% confluency and synchronously infected with Sindbis virus with a translationally fused green fluorescent protein in the nonstructural protein 3 (SINV-nsP3-GFP) at an MOI of 5 PFU per cell. Viral inoculum was allowed to incubate on the cells for 30 min at 4 °C before being replaced with prewarmed media. Bright light and fluorescent microscopy images were taken every 2 h for 30 h. Mean fluorescent count was graphed with standard error mean calculated from 3 replicate wells in the plate with 9 images taken within each well. At the time points indicated, total viral supernatant was removed from the wells and replaced with fresh 1× MEM. Viral supernatant from each time point was then used for plaque assays on BHK-21 cells.
2.4. Virion RNA Extractions, Transfections, and Luciferase Assays
BHK-21 and C7/10 cells were infected with SINV-nLuc, and viral supernatant was obtained at 24 and 48 hpi, respectively. Viral supernatant was purified over a 27% w/v sucrose cushion in HNE buffer for 2.5 h at 43,000× g before being resuspended in HNE buffer. Virion-encapsidated RNA was extracted from the BHK-21- and C7/10-derived viruses (SINV-nsP3-NLuc) using TRiZOL (Sigma Aldrich) following the manufacturer’s protocol. Postextraction, RNAs were DNase (RQ1 RNase-free DNase, NEB)-treated using the manufacturer’s protocol to remove cellular contaminants, and viral RNA copies were quantified via quantitative RT-PCR using primers probing for SINV E1 genomic region and a standard curve comprised of linearized SINV infectious clone containing the full-length viral genome. To determine replication kinetics of Sindbis virion RNA derived from BHK-21 or C7/10 cells, HEK-293 cells were transfected or infected with equal copies of virion-isolated RNA or purified virions (108 copies, quantified using qRT-PCR) in serum-free Opti-MEM (Gibco) using LTX transfection mix (Invitrogen). Transfection was carried out for 4 h before the transfection inoculum was removed and replaced with 1× MEM. Infections were carried out synchronously for 30 min at 4 °C before prewarmed 1× MEM was added. Cells were harvested at indicated times post-transfection and homogenized in 1× Cell Culture Lysis Reagent (Promega). Samples were mixed with NanoGlo luciferase Reagent (Promega), incubated at room temperature for 5 min, then recorded for luminescence using a Synergy H1 microplate reader (BioTech instruments). Total cellular RNA was obtained using the BioRad Aurum total RNA mini kit according to the manufacturer’s protocol. Genomic vRNA copies were then quantified via quantitative RT-PCR using primers probing for SINV nsP1 genomic region to detect only (+) vRNA in the cell and a standard curve comprised of linearized SINV infectious clone containing the full-length viral genome.
2.5. Input RNA Decay
Input RNA Decay assay was adapted from a previously described protocol [
21,
22]. HEK-293 cells were seeded at 80% confluency in a 24-well plate, and 30 min prior to infection, 3× well volume of 1× MEM supplemented with 4SU (Sigma-Aldrich) at a concentration of 50 µM was added to each well. At the time of infection, the 1× MEM supplemented with 4SU was removed and 250 uL of inoculum was added to infect the cells at an MOI of 10 PFU/cell with either BHK-21 derived, or C7/10 derived SINV. The 24-well plate was then placed at 4 °C for 30 min for a synchronous infection of virus. After 30 min, the plate was removed from 4 °C, the inoculum was removed, and prewarmed 1× MEM supplemented with 50 µM 4SU was added to the cells to allow virus to be absorbed. Five minutes later, the media were removed, the cells were washed 3× with PBS to remove any unbound virus, 1× MEM supplemented with 50 µM 4SU was added, and the 0 min postinfection time point was harvested. Time points were harvested at 0, 45, 90, and 135 min postinfection. Media were removed and RNA Lysis Solution was added to each well before being moved to a clean 1.5 mL tube for further RNA extraction using the BioRad Aurum Total RNA mini kit according to the manufacturer’s protocol. Streptavidin agarose beads (ThermoFisher) were incubated with EZ-Link HPDP-Biotin (ThermoFisher) at 10× the binding capacity for 30 min at 25 °C under gentle agitation. After incubation, the biotin-bound beads were washed 3× with PBS before being added to 1 µg of total cellular RNA for each sample. The biotin-bound beads and RNA were incubated for 1.5 h at 20 °C under gentile agitation. Following incubation, the beads were spun down at 6000×
g for 5 min and the unbound, unlabeled RNA supernatant was removed. The unbound RNA was then cleaned up using the RNeasy mini kit (Qiagen) following the manufacturer’s protocol and quantified by Nanodrop to determine RNA concentration. The purified, unlabeled RNA was then used for qRT-PCR to determine relative unlabeled viral RNA at each time point compared to the 0 min postinfection time point. The purified, unlabeled, cellular RNA was then used as a template to synthesize cDNA using M-MuLV Reverse Transcriptase (NEB) with random hexamer primers (Integrated DNA Technologies). Quantitative RT-PCR analysis was performed using Brilliant III SYBR green QPCR master mix (Thomas Scientific) with SINV E1 genomic region-specific primers and Human 18S primers according to the manufacturer’s protocol and with the Applied Bioscience StepOnePlus qRT-PCR machine (Life Technologies). The relative abundance of SINV genomic RNA was determined by normalizing to Human 18S expression using the delta-delta comparative threshold method (ΔΔCT) and compared back to the 0 min postinfection sample.
2.6. Western Blotting
HEK-293 were infected with 108 genomes of purified SINV-nsP3-NLuc derived from either C7/10 or BHK-21 cells, as described previously. At 9 h postinfection, cells were lysed and harvested using Lysis Buffer II (10 mM Tris pH 7.4, 150 mM NaCl, 1% Triton-X100, 10 mM NaH2PO4, 5 mM EDTA, and 1× PMSF). Protein was extracted by vortexing for 10 s every 2 min over 10 min. At the end of 10 min, the samples were vortexed one last time then spun down for 10 min at 10,000 RPM. The supernatant was extracted and moved to a fresh tube. The samples were then mixed with SDS-loading buffer to a concentration of 1× and heated at 95 °C for 5 min. The samples were then run through a 10% SDS-PAGE gel and transferred to a PVDF membrane. The membrane was blocked in 5% TBSM (Tris Buffer Saline 1× + 5% Dry Milk) for 1 h at room temperature. Following blocking, the membrane was incubated with rabbit anti-nsP2 or rabbit anti-β actin at 4 °C overnight. Following incubation with primary antibody, the membrane was washed with TBST prior to incubation with goat anti-rabbit AlexaFluor 750 (ThermoFisher) for 1 h at room temperature. Following secondary antibody incubation, the membrane was imaged using a BioRad ChemiDoc MP Imaging System. Band intensity was determined using ImageStudio Lite Ver. 5.2. Band intensity is represented as a ratio of nsP2 signal to β-actin signal and standardized to the BHK-21-derived virus sample.
2.7. Quantification of RNA Modifications by LC-MS/MS
BHK-21- and C7/10-derived Sindbis virus were produced as explained in Obtaining Viruses. Virion-encapsidated RNA was harvested using TRiZOL (Sigma Aldrich) following the manufacturer’s protocol. Total RNA (~1 µg) was digested by nuclease P1 (10 Units) at 50 °C for 16 h. Additional Tris pH 7.5 was then added to a final concentration of 100 mM to adjust the pH, which was followed by the addition of calf intestinal alkaline phosphatase (CIP, NEB, 2 Units). The mixture was incubated at 37 °C for 1 h to convert nucleotide 5′-monophosophates to their respective nucleosides. A total of 10µL of RNA samples were diluted to 30µL and filtered (0.22 µm pore size). A total of 10 µL of the sample was used for LC-MS/MS. Briefly, nucleosides were separated on a C18 column (Zorbax Eclipse Plus C18 column, 2.1 × 50 mm, 1.8 micron) paired with an Agilent 6490 QQQ Triple-quadrupole LC mass spectrometer using multiple-reaction monitoring in positive-ion mode. The nucleosides were quantified using the retention time of the pure standards and the nucleoside-to-base-ion mass transitions of 268.1 to 136 (A), 244.1 to 113 (C), 284.2 to 152 (G), 300 to 168.1 (8-oxoG), 282.2 to 150 (m1A), 298 to 166 (m1G), 258 to 126 (m3C and m5C), 282.1 to 150 (m6A), 298 to 166 (m7G). Standard calibration curves were generated for each nucleoside by fitting the signal intensities against concentrations of pure nucleoside preparations. The curves were used to determine the concentration of the respective nucleoside in the sample. The A, G, and C standards were purchased from ACROS ORGANICS; m5C was purchased from BioVision; m7G, m1G, and m3C were purchased from Carbosynth; m6G and m6A were purchased from Berry’s Associates; and m1A was purchased from Cayman Chemical Company. The modification level on the nucleosides was calculated as the ratio of modified: unmodified.
2.8. In Vitro Translation
BHK-21 and C7/10 cells were infected with SINV-nLuc, and viral supernatant was obtained at 24 and 48 hpi, respectively. Viral supernatant was purified over a 27% w/v sucrose cushion in HNE buffer for 2.5 h at 43,000× g before being resuspended in HNE buffer. Virion-encapsidated RNA was extracted from the BHK-21- and C7/10-derived viruses (SINV-nsP3-NLuc) using TRiZOL (Sigma Aldrich) following the manufacturer’s protocol. Postextraction, RNAs were DNase (RQ1 RNase-free DNase, NEB)-treated using the manufacturer’s protocol to remove cellular contaminants, and viral RNA copies were quantified via quantitative RT-PCR using primers probing for SINV E1 genomic region and a standard curve comprised of linearized SINV infectious clone containing the full-length viral genome. Rabbit Reticulocyte Lysate, Nuclease-Treated (Promega), was used according to the manufacturer’s protocol. Briefly, 108 genomes of BHK-21- and C7/10-derived purified vRNA was added to the Rabbit Reticulocyte Lysate and incubated at 30 °C for 90 min. A plasmid containing the full-length genome of SINV-nsP3-NLuc was used as a template for in vitro transcription using MEGAscript SP6 Transcription Kit (ThermoFisher) following the manufacturer’s protocol. As a control, 2 µG of the in vitro transcribed RNA was added to the Rabbit Reticulocyte Lysate following the manufacturer’s protocol. Following incubation, 5 µL of the in vitro translation was added to 25 µL of NanoGlo luciferase Reagent (Promega), incubated at room temperature for 5 min then recorded for luminescence using a Synergy H1 microplate reader (BioTech instruments).
2.9. Statistical Analysis of Experimental Data
All statistical analyses were performed using GraphPad Prism 9 (GraphPad Software Inc., San Diego, CA, USA).
2.10. Graphics
Graphical assets made in ©BioRender—
biorender.com. Accessed on 25 August 2022.
4. Discussion
Arboviruses are defined by their capacity to replicate and infect both vertebrate and nonvertebrate hosts in a cyclic infection cycle. Understanding the differences in nonvertebrate- and vertebrate-derived arboviruses and how those differences affect replication is an important step in figuring out how to counteract these viruses. While there is evidence that host origin influences alphavirus infectivity, there is much still to be explored about what happens to the virus after replication in vertebrate or nonvertebrate hosts that primes it for subsequent host infection [
2,
3,
4,
5,
6]. RNA modifications of viral RNA is a newly emerging field in virology. While modification of vRNA was identified as early as the 1970s, it is only recently that the effect of those modifications on viral replication has begun to be explored [
27,
28,
29]. N6-methyladenosine (m6A) has previously been shown to influence flavivirus replication in mammalian cells, and 5-methylcytosine (m5C) modifications have been shown to influence alphavirus replication in arthropod cells [
14,
15]. The effects of RNA modifications on the Sindbis virus specifically have not been fully explored, and to that affect, the differences of viral RNA modifications from different host origins have not been examined. This information leads us to ask the following question in this present study: are the differences in replication efficiency of viruses derived from mosquito and mammalian hosts related to the RNA modification state of virion genomic RNA?
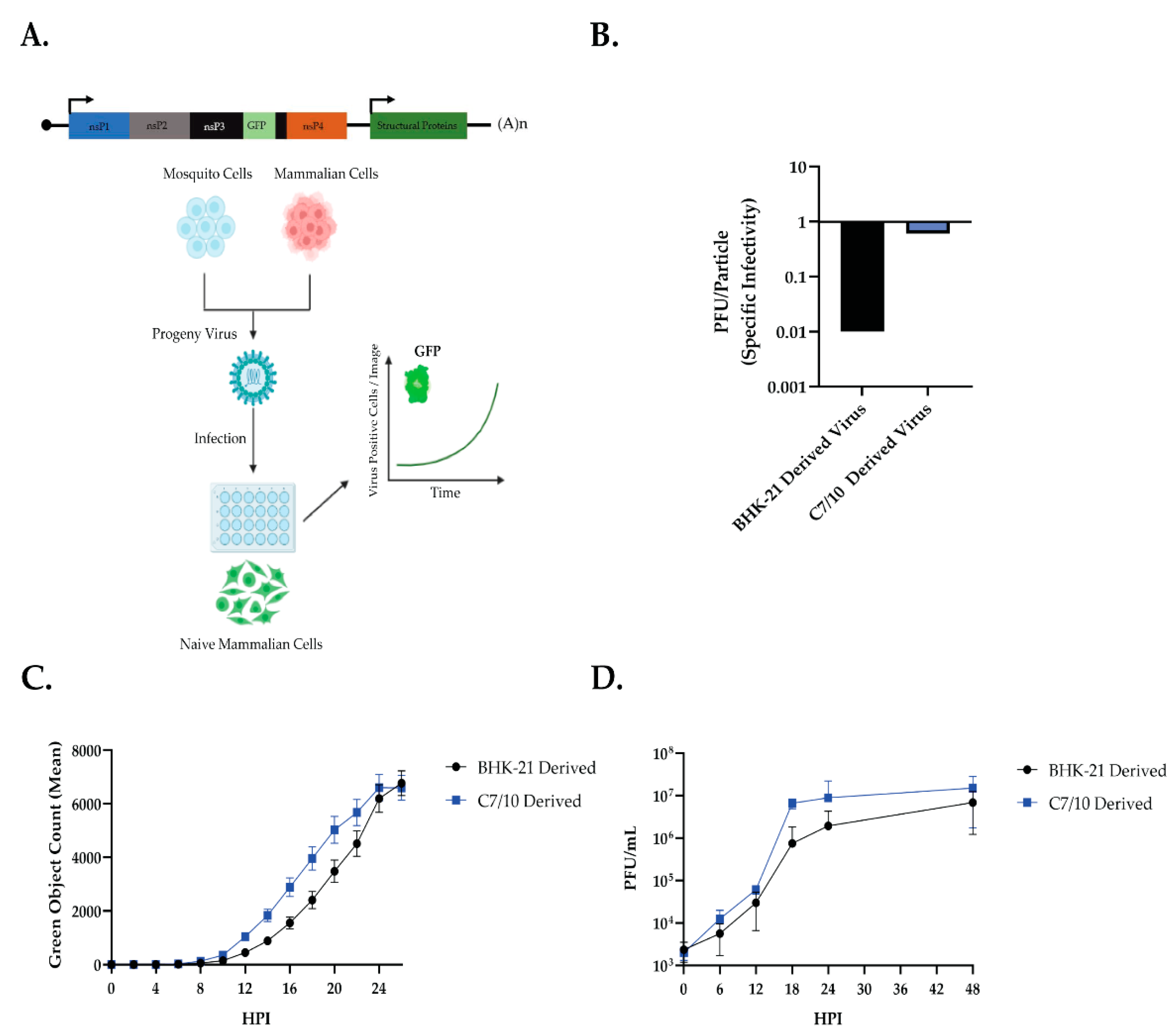
Mosquito-derived SINV grows faster and produces more infectious viruses than mammalian-derived SINV in HEK-293 cells (
Figure 1). It has been well documented that mosquito-derived arboviruses tend to grow better on mammalian cells than mammalian-derived, and vice versa [
24,
30]. We have shown that this effect is similar using SINV to infect HEK-293 cells. We observed an early advantage of the mosquito-derived SINV, however, which we decided to explore further (
Figure 1C,D).
Luciferase assays showed that mosquito-derived SINV significantly increased translation early in infection (
Figure 2B). However, the luciferase assay alone is not enough to conclude whether the early advantage of the mosquito-derived virus is due to translation alone. The increased translation as measured by luciferase assay could be due to increased amounts of genomic vRNA in the cell, which could be due to either the incoming genomic vRNA being more stable, or that the RNA levels are higher. To address these possibilities for the advantage, we looked at both RNA stability having an effect and increased replication of the viral genome (
Figure 2C,D). We observed no significant difference in the amount of the genomic viral RNA in the cell infected with the mosquito-derived SINV (~2.5-fold increase) and a small but significant increase in the amount of extracellular vRNA (~1.18-fold increase) (
Figure 2D). However, when we measured the amount of luciferase signal as a ratio of genomic vRNA we found a significant increase in the mosquito-derived virus, ~5.5-fold (
Figure 2D, far right). While there appears to be a slight increase in the amount of vRNA present in the mosquito-derived virus infection, the relative amount of luciferase produced per genome shows that the mosquito-derived genomic RNA seems to be a better template for translation. Further supporting this observation was the demonstration of an increased amount of nsP2 produced in the cells infected with mosquito-derived virus (
Figure 2E), leading us to the conclusion that the differences in RNA modifications are influencing the translational efficiency of the RNA. The results observed in
Figure 2 are supported by the ability of purified vRNA alone being sufficient to phenocopy what is seen with intact virus particles, as we see both a significant increase in RLU from purified mosquito-derived genomic RNA transfected into HEK-293 cells, as well as in the in vitro translation assay (
Figure 3B,C). Surprisingly, there was no significant difference in the stability of the incoming genomic RNA from mosquito or mammalian-derived viruses, as we know that RNA modifications can affect the stability of some RNAs [
10,
16,
17] (
Figure 2C). These results support the hypothesis that acquired RNA modifications from different hosts are also influencing SINV infection, specifically by increasing the translational efficiency of the vRNA, which in turn leads to more infectious viruses being produced. This, along with other known host-dependent molecular modifications of arboviruses, further expand our understanding of the impact of replication background on viral replication and transmission.
The effect of RNA modifications on viral RNA is a newly emerging field with evidence already showing the impact RNA modifications have on the viral life cycle [
9,
13,
14,
15,
19,
20]. There is little known about the effects, let alone the presence, of RNA modifications on alphaviruses specifically or if those modifications are different depending on the host the virus is derived from. We show using liquid chromatography in tandem with mass spectrometry that mosquito-derived SINV and mammalian-derived SINV have significantly different levels of varying RNA modifications. For instance, we see that mosquito-derived SINV s significantly more m5C-modified than the mammalian counterpart, and the opposite is true for m6A modification. This is an interesting find, as previous studies have shown that both m5C and m6A modifications have an impact on arboviruses. More specifically, m5C has been shown to play an important role in the pathogen-blocking ability of
Wolbachia in insect cells, and m6A modifications have been more extensively studied in the context of mammalian cells [
9,
13,
14,
15,
19,
20]. The difference in the number of m6A modifications in mosquito- and mammalian-derived viruses is consequential for a few reasons: (i) it is in accord with previous data that show that m6A modifications play a role in mammalian cell systems for other arboviruses, and (ii) there is evidence to suggest that m6A modification can help the virus to evade the immune response by escaping RIG-I sensor recognition, which is operative in mammalian cells but not mosquito cells [
14,
27].
Overall, our results suggest that the RNA modifications acquired by the virus in mosquito cells may play a role in translational efficiency of the viral genome in the mammalian cell. This increased translational efficiency and subsequent increase in infectious virus production is likely influenced by the specific modifications acquired in the mosquito. Further studies will try to identify exactly where these modifications are occurring and look to attribute specific nucleotide modifications to the observed phenotypes. It is likely that each individual type of RNA modification is playing a collective role in the observed phenotypes. While the differences in m5C and m6A modifications are interesting based on the amount of previous research and what is known about these modifications’ effects on arbovirus life cycles, the significant differences in the other observed RNA modifications are just as interesting. While the effects of internal m7G, m3C, m1G, m1A, and Ψ modifications are less well known, the observation of significant differences in some of these modifications implies they also play a role in the viral life cycle, though it is currently difficult to say what role that is. Since each of these modifications affect the base pairing of the modified nucleotide, it is tempting to speculate that they may be found in regions of regulation in which secondary structure is important, and may play important roles in maintaining the appropriate stability of RNA structure under host-specific temperature conditions.
RNA modifications in genomic viral RNA can influence a multitude of things, including—but not limited to—RNA stability, RNA localization, and translational efficiency. Another confounding variable is the difficulty in studying single RNA modifications’ effects, as one modification alone may not be enough to elicit any phenotype. Along those lines, how these RNA modifications interact is not well understood and the possibility of epistatic interactions increases the complexity of analysis and interpretation of biological consequences. Finally, the effect of methylation readers needs to be explored. While there is strong evidence for m6A readers, specifically the YTH family of proteins, having an impact on flavivirus RNA function, there is no reported evidence of the effect readers have on SINV or other alphaviruses [
14]. This study opens the door to further studies of the effects that RNA modifications have on arboviruses derived from their arthropod or vertebrate hosts and how those modifications affect RNA function.


{kind=link}
{kind=link}
{kind=link}
{kind=link}



