
Changes in the Cervical Microbiota of Women with Different High-Risk Human Papillomavirus Loads
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population and Ethical Considerations
2.2. Molecular Detection and Quantification of hrHPV
2.3. Illumina Sequencing and Bioinformatics Analyses
2.4. Descriptive and Statistical Analyses of the Bacterial Communities
2.5. Analyses of Differentially Present Genera and Correlation Network Construction
3. Results
3.1. Population Characteristics
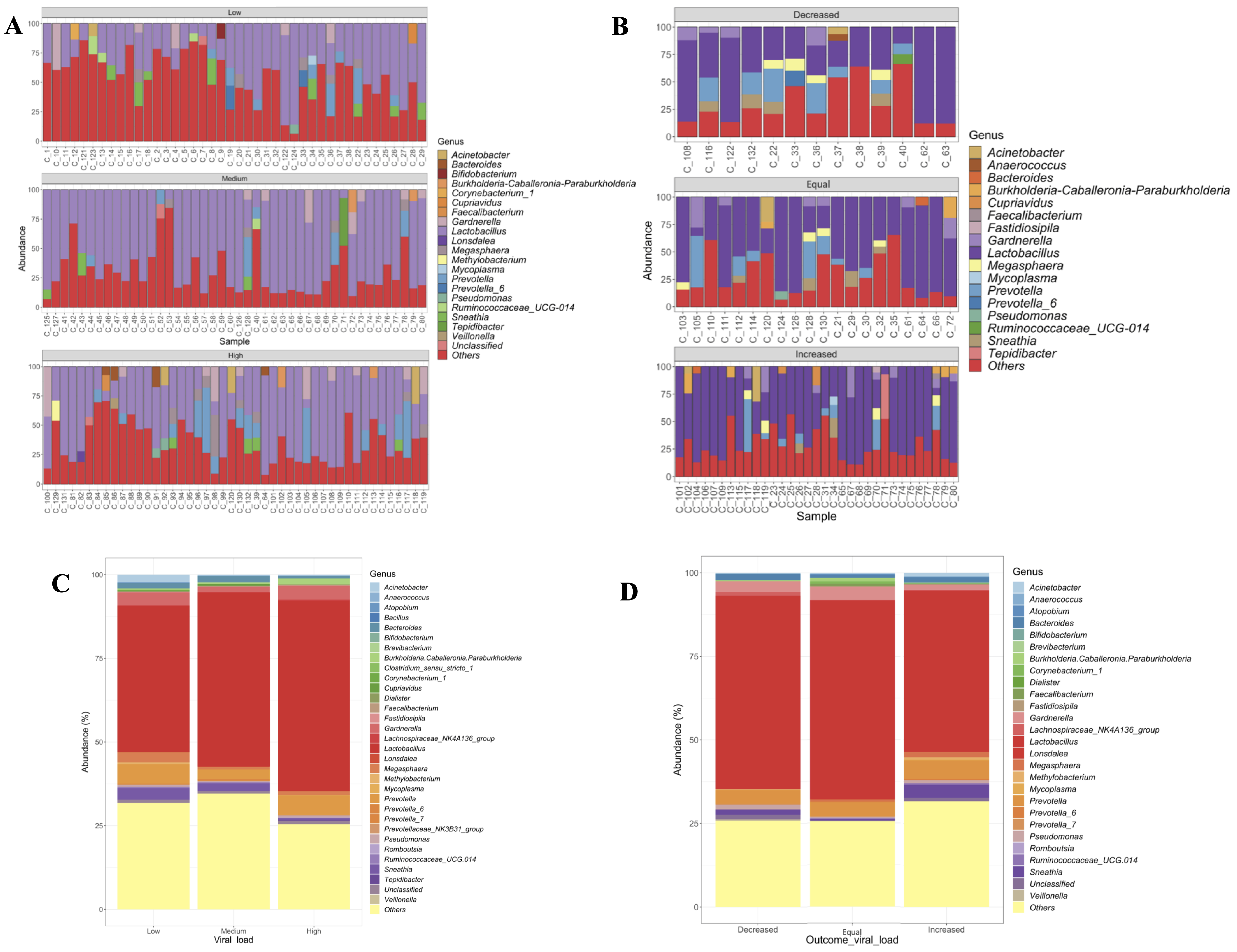
3.2. Cervical Microbiota Composition
3.3. Compositional Differences between Groups
3.4. Alpha and Beta Diversity Analyses
3.5. Identification of Markers and Correlations between Bacterial Communities among the Groups Evaluated
3.6. Lactobacillus and Community State Type Distribution
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Christensen, N.D. HPV disease transmission protection and control. Microb. Cell 2016, 3, 476–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vyshenska, D.; Lam, K.C.; Shulzhenko, N.; Morgun, A. Interplay between viruses and bacterial microbiota in cancer development. Semin. Immunol. 2017, 32, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Boda, D.; Docea, A.O.; Calina, D.; Ilie, M.A.; Caruntu, C.; Zurac, S.; Neagu, M.; Constantin, C.; Branisteanu, D.E.; Voiculescu, V.; et al. Human papilloma virus: Apprehending the link with carcinogenesis and unveiling new research avenues (Review). Int. J. Oncol. 2018, 52, 637–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Río-Ospina, D.; Camargo, M.; León, S.D.; Sara, C.; Sánchez, R.; Moreno-Pérez, D.A.; Patarroyo, M.E.; Patarroyo, M.A. Identifying the HLA DRB1-DQB1 molecules and predicting epitopes associated with high-risk HPV infection clearance and redetection. Sci. Rep. 2020, 10, 7306. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Sekine, M.; Hanley, S.J.B.; Kudo, R.; Hara, M.; Adachi, S.; Ueda, Y.; Miyagi, E.; Enomoto, T. Risk factors for HPV infection and high-grade cervical disease in sexually active Japanese women. Sci. Rep. 2021, 11, 2898. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.Y.; Kim, M.K.; Seo, S.; Lee, D.O.; Chung, Y.K.; Lim, M.C.; Kim, J.; Lee, C.W.; Park, S. Alcohol consumption and persistent infection of high-risk human papillomavirus. Epidemiol. Infect. 2015, 143, 1442–1450. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, A.; Yang, J.; Zhao, W.; Wang, Z.; Wang, W.; Wang, J.; Song, J.; Li, L.; Lv, W.; et al. Dietary nutrient intake related to higher grade cervical intraepithelial neoplasia risk: A Chinese population-based study. Nutr. Metab. 2020, 17, 100. [Google Scholar] [CrossRef]
- Oyervides-Muñoz, M.A.; Pérez-Maya, A.A.; Sánchez-Domínguez, C.N.; Berlanga-Garza, A.; Antonio-Macedo, M.; Valdéz-Chapa, L.D.; Cerda-Flores, R.M.; Trevino, V.; Barrera-Saldaña, H.A.; Garza-Rodríguez, M.L. Multiple HPV Infections and Viral Load Association in Persistent Cervical Lesions in Mexican Women. Viruses 2020, 2, 380. [Google Scholar] [CrossRef] [Green Version]
- Silva, J.; Cerqueira, F.; Medeiros, R. Chlamydia trachomatis infection: Implications for HPV status and cervical cancer. Arch. Gynecol. Obstet. 2014, 289, 715–723. [Google Scholar] [CrossRef]
- Carmona-Gutierrez, D.; Kainz, K.; Madeo, F. Sexually transmitted infections: Old foes on the rise. Microb. Cell 2016, 3, 361–362. [Google Scholar] [CrossRef]
- Audirac-Chalifour, A.; Torres-Poveda, K.; Bahena-Román, M.; Téllez-Sosa, J.; Martínez-Barnetche, J.; Cortina-Ceballos, B.; López-Estrada, G.; Delgado-Romero, K.; Burguete-García, A.I.; Cantú, D.; et al. Cervical Microbiome and Cytokine Profile at Various Stages of Cervical Cancer: A Pilot Study. PLoS ONE 2016, 11, e0153274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- León, S.D.; Sara, C.; Río-Ospina, D.; Camargo, M.; Sánchez, R.; Moreno-Pérez, D.A.; Pérez-Prados, A.; Patarroyo, M.E.; Patarroyo, M.A. Persistence, clearance and reinfection regarding six high risk human papillomavirus types in Colombian women: A follow-up study. BMC Infect. Dis. 2014, 14, 395. [Google Scholar]
- Di Paola, M.; Sani, C.; Clemente, A.M.; Iossa, A.; Perissi, E.; Castronovo, G.; Tanturli, M.; Rivero, D.; Cozzolino, F.; Cavalieri, D.; et al. Characterization of cervico-vaginal microbiota in women developing persistent high-risk Human Papillomavirus infection. Sci. Rep. 2017, 7, 10200. [Google Scholar] [CrossRef]
- Usyk, M.; Zolnik, C.P.; Castle, P.E.; Porras, C.; Herrero, R.; Gradissimo, A.; Gonzalez, P.; Safaeian, M.; Schiffman, M.R.; Burk, R.D.; et al. Cervicovaginal microbiome and natural history of HPV in a longitudinal study. PLoS Pathog. 2020, 16, e1008376. [Google Scholar] [CrossRef] [Green Version]
- Ravel, J.; Gajer, P.; Abdo, Z.; Schneider, G.M.; Koenig, S.S.; McCulle, S.L.; Karlebach, S.; Gorle, R.; Russell, J.; Tacket, C.O.; et al. Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. USA 2011, 108, 4680–4687. [Google Scholar] [CrossRef] [Green Version]
- Mitra, A.; MacIntyre, D.A.; Marchesi, J.R.; Lee, Y.S.; Bennett, P.R.; Kyrgiou, M. The vaginal microbiota, human papillomavirus infection and cervical intraepithelial neoplasia: What do we know and where are we going next? Microbiome 2016, 4, 58. [Google Scholar] [CrossRef] [Green Version]
- Mitra, A.; MacIntyre, D.A.; Lee, Y.S.; Smith, A.; Marchesi, J.R.; Lehne, B.; Bhatia, R.; Lyons, D.; Paraskevaidis, E.; Li, J.V.; et al. Cervical intraepithelial neoplasia disease progression is associated with increased vaginal microbiome diversity. Sci. Rep. 2015, 5, 16865. [Google Scholar] [CrossRef] [Green Version]
- Gillet, E.; Meys, J.F.; Verstraelen, H.; Bosire, C.; De Sutter, P.; Temmerman, M.; Broeck, D.V. Bacterial vaginosis is associated with uterine cervical human papillomavirus infection: A meta-analysis. BMC Infect. Dis. 2011, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- Kyrgiou, M.; Mitra, A.; Moscicki, A.B. Does the vaginal microbiota play a role in the development of cervical cancer? Transl. Res. 2017, 179, 168–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Río-Ospina, D.; León, S.D.; Cecilia, S.; Camargo, M.; Moreno-Pérez, D.A.; Sánchez, R.; Pérez-Prados, A.; Patarroyo, M.E.; Patarroyo, M.A. The DNA load of six high-risk human papillomavirus types and its association with cervical lesions. BMC Cancer 2015, 15, 100. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.E.; Lee, S.; Lee, H.; Song, Y.-M.; Lee, K.; Han, M.J.; Sung, J.; Ko, G. Association of the vaginal microbiota with human papillomavirus infection in a Korean twin cohort. PLoS ONE 2013, 8, e63514. [Google Scholar] [CrossRef]
- Lee, J.E.; Lee, S.; Lee, H.; Song, Y.-M.; Lee, K.; Han, M.J.; Sung, J.; Ko, G. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. S1), 4516–4522. [Google Scholar]
- D'Argenio, V.; Casaburi, G.; Precone, V.; Salvatore, F. Comparative metagenomic analysis of human gut microbiome composition using two different bioinformatic pipelines. BioMed Res. Int. 2014, 2014, 325340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Gonzalez Peña, A.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callahan, B. Silva Taxonomic Training Data Formatted for DADA2 (Silva Version 132). 2018. Available online: https://zenodo.org/record/1172783#.Y3jW4UlBy3A (accessed on 15 November 2022).
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Wickham, H. Reshaping Data with the reshape Package. J. Stat. Softw. 2007, 21, 1–20. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Weiss, S.; Xu, Z.Z.; Peddada, S.; Amir, A.; Bittinger, K.; Gonzalez, A.; Lozupone, C.; Zaneveld, J.R.; Vázquez-Baeza, Y.; Birmingham, A.; et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 2017, 5, 27. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Hong, Z.; Wang, W.; Gu, L.; Gao, H.; Qiu, L.; Di, W. Association between the vaginal microbiome and high-risk human papillomavirus infection in pregnant Chinese women. BMC Infect. Dis. 2019, 19, 677. [Google Scholar] [CrossRef]
- van Duin, M.; Snijders, P.J.; Schrijnemakers, H.F.; Voorhorst, F.J.; Rozendaal, L.; Nobbenhuis, M.A.; van den Brule, A.J.; Verheijen, R.H.; Helmerhorst, T.J.; Meijer, C.J. Human papillomavirus 16 load in normal and abnormal cervical scrapes: An indicator of CIN II/III and viral clearance. Int. J. Cancer 2002, 98, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Zuo, J.; Huang, Y.; An, J.; Yang, X.; Li, N.; Huang, M.; Wu, L. Nomograms based on HPV load for predicting survival in cervical squamous cell carcinoma: An observational study with a long-term follow-up. Chin. J. Cancer Res. 2019, 31, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Constandinou-Williams, C.; Collins, S.I.; Roberts, S.; Young, L.S.; Woodman, C.B.; Murray, P.G. Is human papillomavirus viral load a clinically useful predictive marker? A longitudinal study. Cancer Epidemiol. Biomark. Prev. 2010, 19, 832–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehlner-Gardiner, C.; Roshick, C.; Carlson, J.H.; Hughes, S.; Belland, R.J.; Caldwell, H.D.; McClarty, G. Molecular basis defining human Chlamydia trachomatis tissue tropism. A possible role for tryptophan synthase. J. Biol. Chem. 2002, 277, 26893–26903. [Google Scholar] [CrossRef] [Green Version]
- Sherchand, S.P.; Ibana, J.A.; Zea, A.H.; Quayle, A.J.; Aiyar, A. The High-Risk Human Papillomavirus E6 Oncogene Exacerbates the Negative Effect of Tryptophan Starvation on the Development of Chlamydia trachomatis. PLoS ONE 2016, 11, e0163174. [Google Scholar] [CrossRef] [Green Version]
- Ziklo, N.; Vidgen, M.E.; Taing, K.; Huston, W.M.; Timms, P. Dysbiosis of the Vaginal Microbiota and Higher Vaginal Kynurenine/Tryptophan Ratio Reveals an Association with Chlamydia trachomatis Genital Infections. Front. Cell Infect. Microbiol. 2018, 8, 1. [Google Scholar] [CrossRef] [Green Version]
- Chintala, S.; Levan, J.; Robinson, K.; Quist, K.; Katzenellenbogen, R.A. Genes Regulated by HPV 16 E6 and High Expression of NFX1-123 in Cervical Cancers. Onco-Targets Ther. 2020, 13, 6143–6156. [Google Scholar] [CrossRef]
- Boskey, E.R.; Telsch, K.M.; Whaley, K.J.; Moench, T.R.; Cone, R.A. Acid production by vaginal flora in vitro is consistent with the rate and extent of vaginal acidification. Infect. Immun. 1999, 67, 5170–5175. [Google Scholar] [CrossRef] [Green Version]
- Molenaar, M.C.; Singer, M.; Ouburg, S. The two-sided role of the vaginal microbiome in Chlamydia trachomatis and Mycoplasma genitalium pathogenesis. J. Reprod. Immunol. 2018, 130, 11–17. [Google Scholar] [CrossRef]
- Witkin, S.S.; Mendes-Soares, H.; Linhares, I.M.; Jayaram, A.; Ledger, W.J.; Forney, L.J. Influence of vaginal bacteria and D- and L-lactic acid isomers on vaginal extracellular matrix metalloproteinase inducer: Implications for protection against upper genital tract infections. mBio 2013, 4, e00460. [Google Scholar] [CrossRef] [Green Version]
- Zheng, N.; Guo, R.; Wang, J.; Zhou, W.; Ling, Z. Contribution of Lactobacillus iners to Vaginal Health and Diseases: A Systematic Review. Front. Cell Infect. Microbiol. 2021, 11, 792787. [Google Scholar] [CrossRef] [PubMed]
- Petrova, M.I.; Reid, G.; Vaneechoutte, M.; Lebeer, S. Lactobacillus iners: Friend or Foe? Trends Microbiol. 2017, 25, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Sugimura, N.; Li, Q.; Chu, E.S.H.; Lau, H.C.H.; Fong, W.; Liu, W.; Liang, C.; Nakatsu, G.; Su, A.C.Y.; Coker, O.O.; et al. Lactobacillus gallinarum modulates the gut microbiota and produces anti-cancer metabolites to protect against colorectal tumourigenesis. Gut 2021, 71, 2011–2021. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, J.; Han, Q.; Chu, W.; Lu, G.; Chan, W.-Y.; Qin, Y.; Du, Y. in the vaginal microbiota associated with primary ovarian failure. BMC Microbiol. 2020, 20, 230. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Weng, J.; Gao, Y.; Chen, X. Comparison of the vaginal microbiota diversity of women with and without human papillomavirus infection: A cross-sectional study. BMC Infect. Dis. 2013, 13, 271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, V.K.; Paul, S.; Dutta, C. Geography, Ethnicity or Subsistence-Specific Variations in Human Microbiome Composition and Diversity. Front. Microbiol. 2017, 8, 1162. [Google Scholar] [CrossRef] [Green Version]
- Das Purkayastha, S.; Bhattacharya, M.K.; Prasad, H.K.; Upadhyaya, H.; Lala, S.D.; Pal, K.; Das, M.; Sharma, G.D.; Bhattacharjee, M.J. Contrasting diversity of vaginal lactobacilli among the females of Northeast India. BMC Microbiol. 2019, 19, 198. [Google Scholar] [CrossRef] [Green Version]
- Borgdorff, H.; Van Der Veer, C.; Van Houdt, R.; Alberts, C.J.; De Vries, H.J.; Bruisten, S.M.; Snijder, M.B.; Prins, M.; Geerlings, S.E.; van der Loeff, M.F.S.; et al. The association between ethnicity and vaginal microbiota composition in Amsterdam, the Netherlands. PLoS ONE 2017, 12, e0181135. [Google Scholar] [CrossRef] [Green Version]
- Kato, I.; Zhang, J.; Sun, J. Bacterial-Viral Interactions in Human Orodigestive and Female Genital Tract Cancers: A Summary of Epidemiologic and Laboratory Evidence. Cancers 2022, 14, 425. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Univariate Analysis | Multivariate Analysis | ||||
|---|---|---|---|---|---|---|
| OR | 95%CI | p-Value | ORa 1 | 95%CI | p-Value | |
| Viral Load | ||||||
| Low | Reference | Reference | ||||
| Medium | 0.13 | 0.03–0.45 | 0.001 | 0.12 | 0.03–0.44 | 0.001 |
| High | 0.22 | 0.07–0.73 | 0.013 | 0.23 | 0.07–0.78 | 0.018 |
| Viral Load Outcome | ||||||
| Decrease | Reference | Reference | ||||
| Equal | 0.64 | 0.23–1.79 | 0.403 | 0.58 | 0.20–1.64 | 0.312 |
| Increase | 1.05 | 0.40–2.75 | 0.907 | 0.93 | 0.35–2.46 | 0.892 |
| Variable | Univariate Analysis | Multivariate Analysis | ||||
|---|---|---|---|---|---|---|
| OR | 95%CI | p-Value | ORa 1 | 95%CI | p-Value | |
| Second | Reference | Reference | ||||
| Viral Load | ||||||
| Low | 4.56 | 0.85–6.41 | 0.076 | 6.06 | 1.06–4.55 | 0.042 |
| Medium | 2.07 | 0.39–10.89 | 0.387 | 2.35 | 0.44–12.5 | 0.316 |
| High | Reference | Reference | ||||
| Viral Load Outcome | ||||||
| Decrease | 0.85 | 0.17–4.15 | 0.843 | 0.88 | 0.18–4.35 | 0.881 |
| Equal | 0.90 | 0.21–3.82 | 0.897 | 0.96 | 0.22–4.12 | 0.967 |
| Increase | Reference | Reference | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Camargo, M.; Vega, L.; Muñoz, M.; Sánchez, R.; Patarroyo, M.E.; Ramírez, J.D.; Patarroyo, M.A. Changes in the Cervical Microbiota of Women with Different High-Risk Human Papillomavirus Loads. Viruses 2022, 14, 2674. https://doi.org/10.3390/v14122674
Camargo M, Vega L, Muñoz M, Sánchez R, Patarroyo ME, Ramírez JD, Patarroyo MA. Changes in the Cervical Microbiota of Women with Different High-Risk Human Papillomavirus Loads. Viruses. 2022; 14(12):2674. https://doi.org/10.3390/v14122674
Chicago/Turabian StyleCamargo, Milena, Laura Vega, Marina Muñoz, Ricardo Sánchez, Manuel Elkin Patarroyo, Juan David Ramírez, and Manuel Alfonso Patarroyo. 2022. "Changes in the Cervical Microbiota of Women with Different High-Risk Human Papillomavirus Loads" Viruses 14, no. 12: 2674. https://doi.org/10.3390/v14122674