
Exploration of Potent Antiviral Phytomedicines from Lauraceae Family Plants against SARS-CoV-2 Main Protease

 ,
,  , , and
, , and 
Abstract
:1. Introduction
2. Methodology
2.1. Receptor and Ligand Collection
2.2. Structure-Based Virtual Screening and Re-Docking Simulation
2.3. Molecular Dynamics Simulation
2.4. Essential Dynamics and Dynamic Cross-Correlation Matrix (DCCM) Profiling
3. Results and Discussion
3.1. Structure-Based Virtual Screening
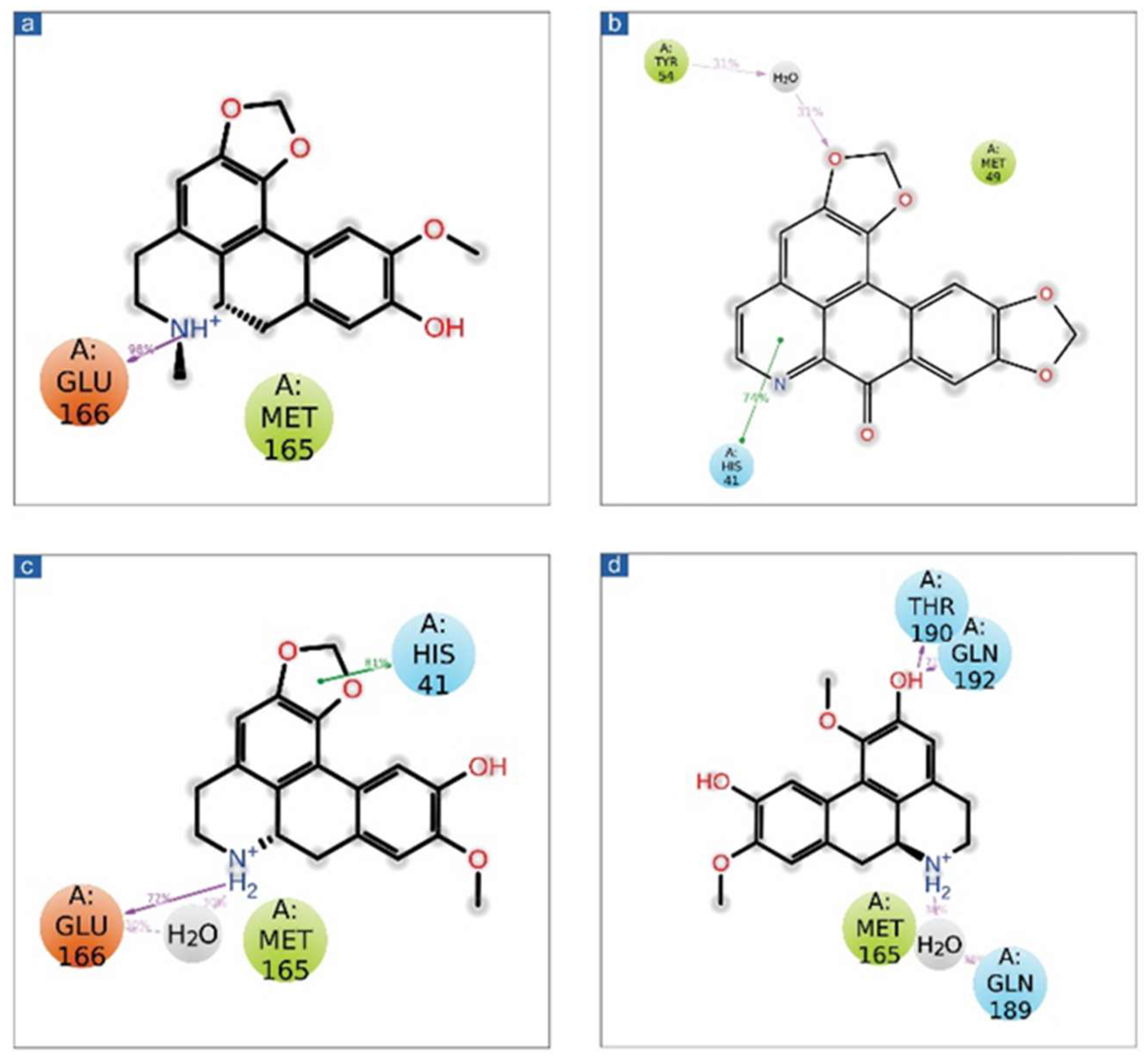
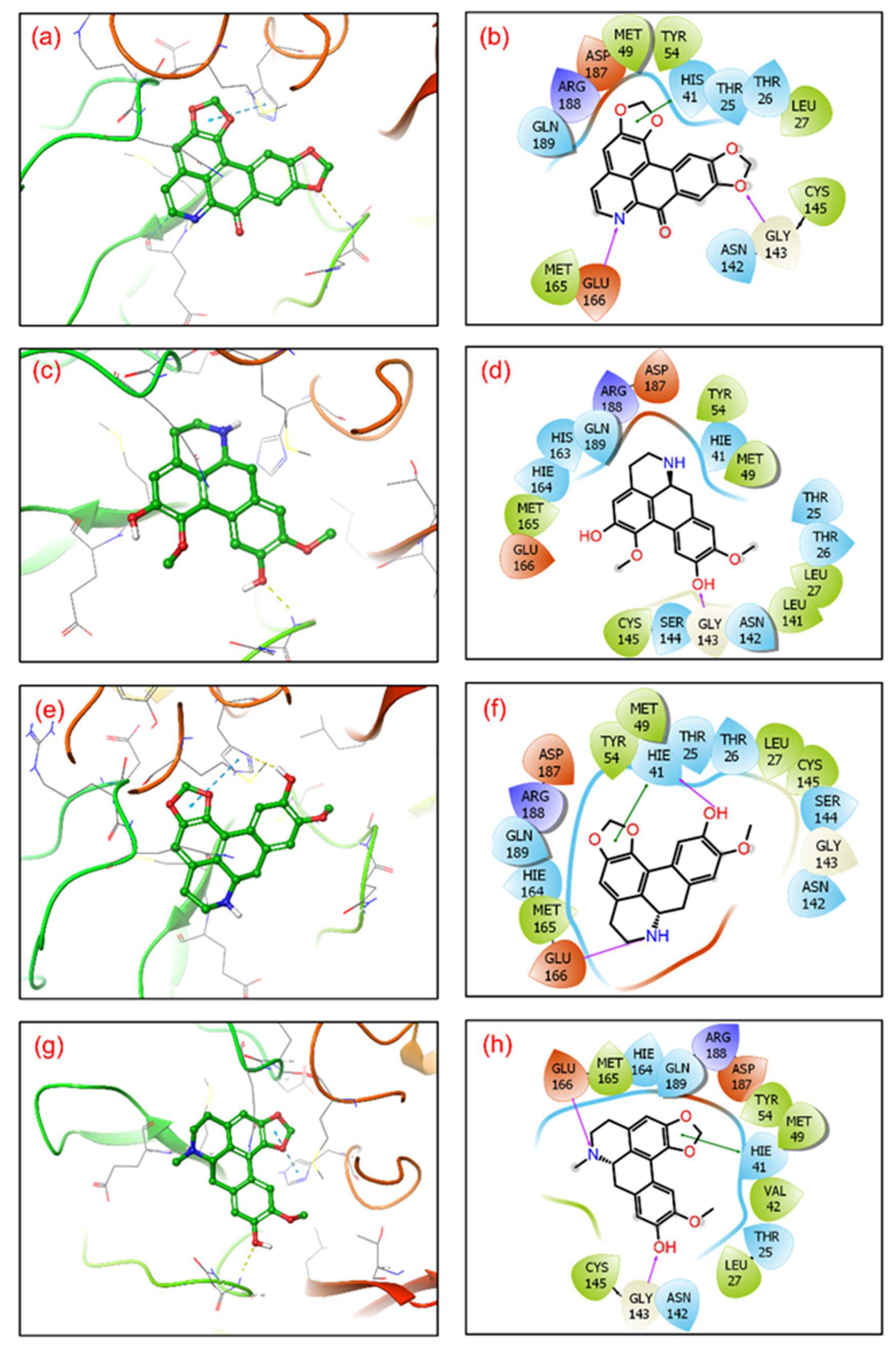
3.2. Re-Docking and Intermolecular Interaction Analysis
3.3. Molecular Dynamics Simulation Analysis
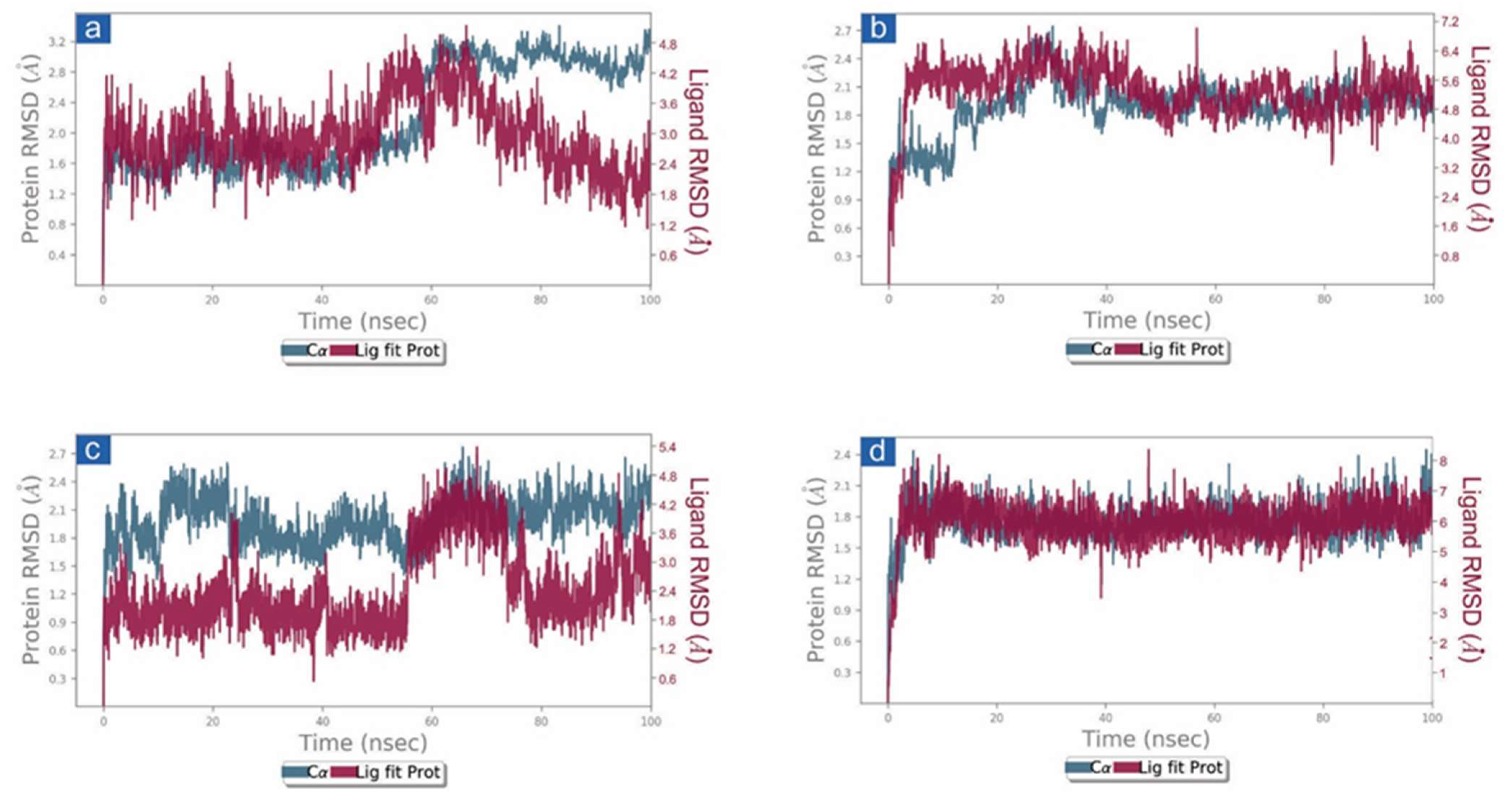
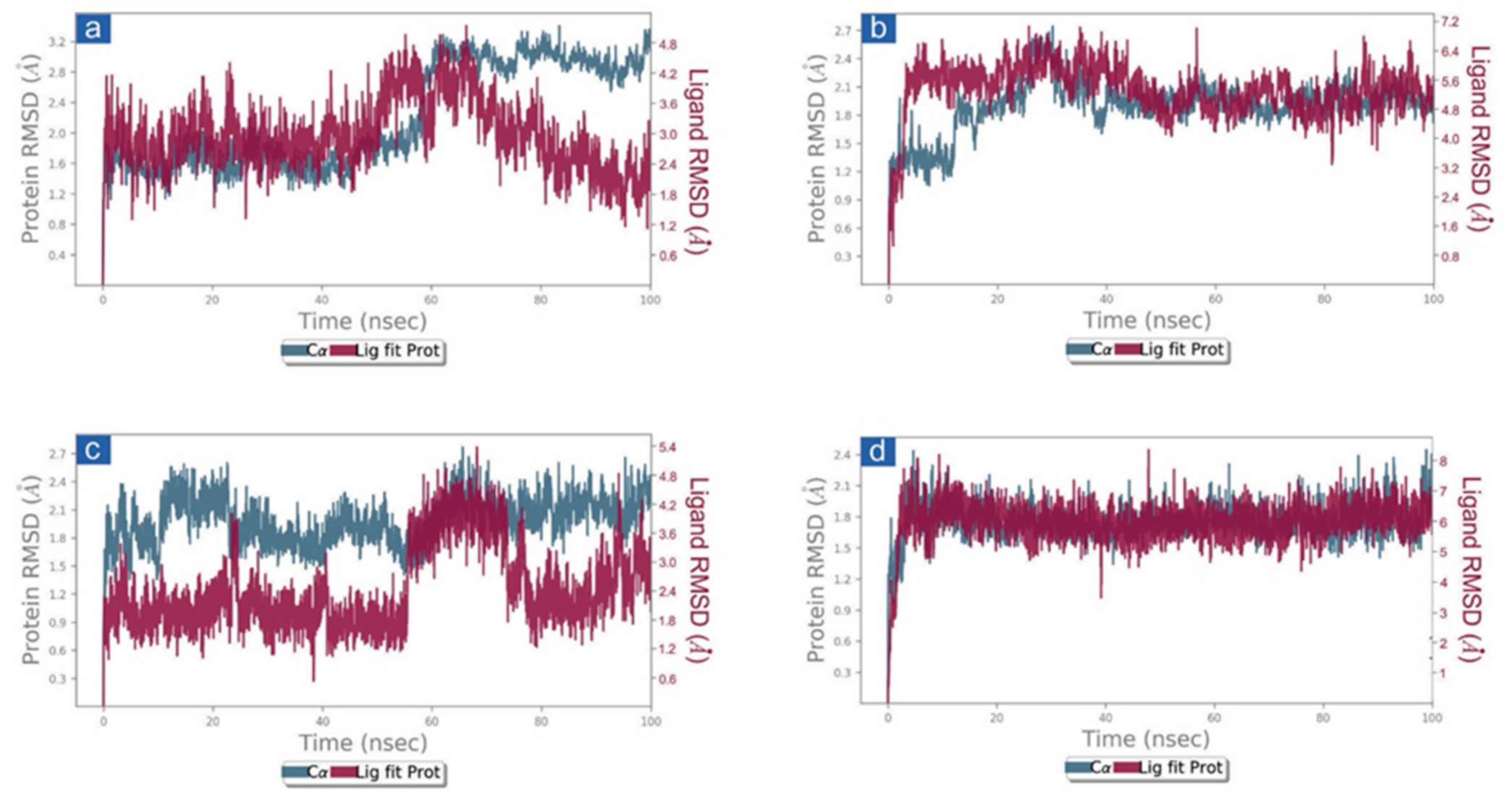
3.3.1. RMSD and RMSF Analysis
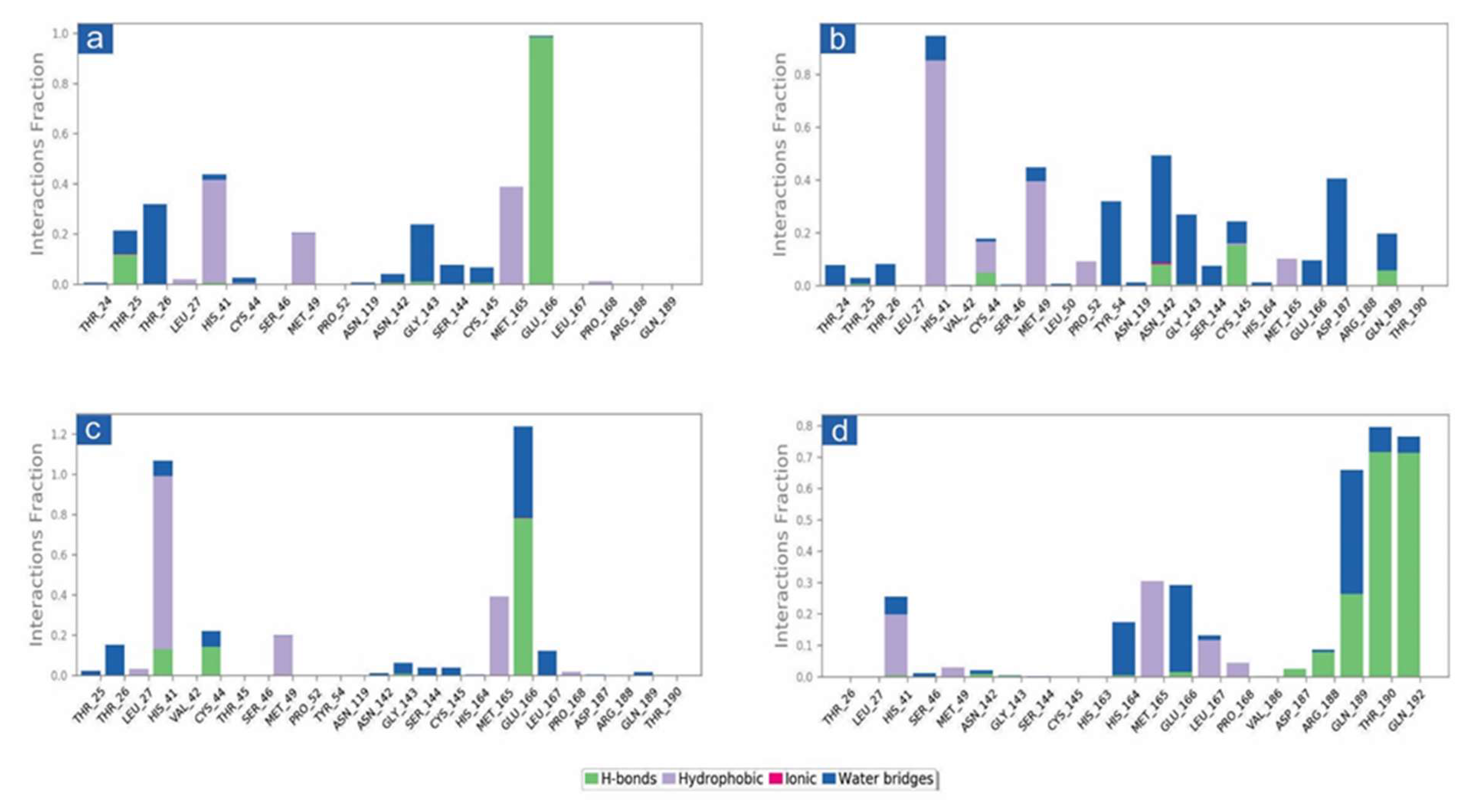
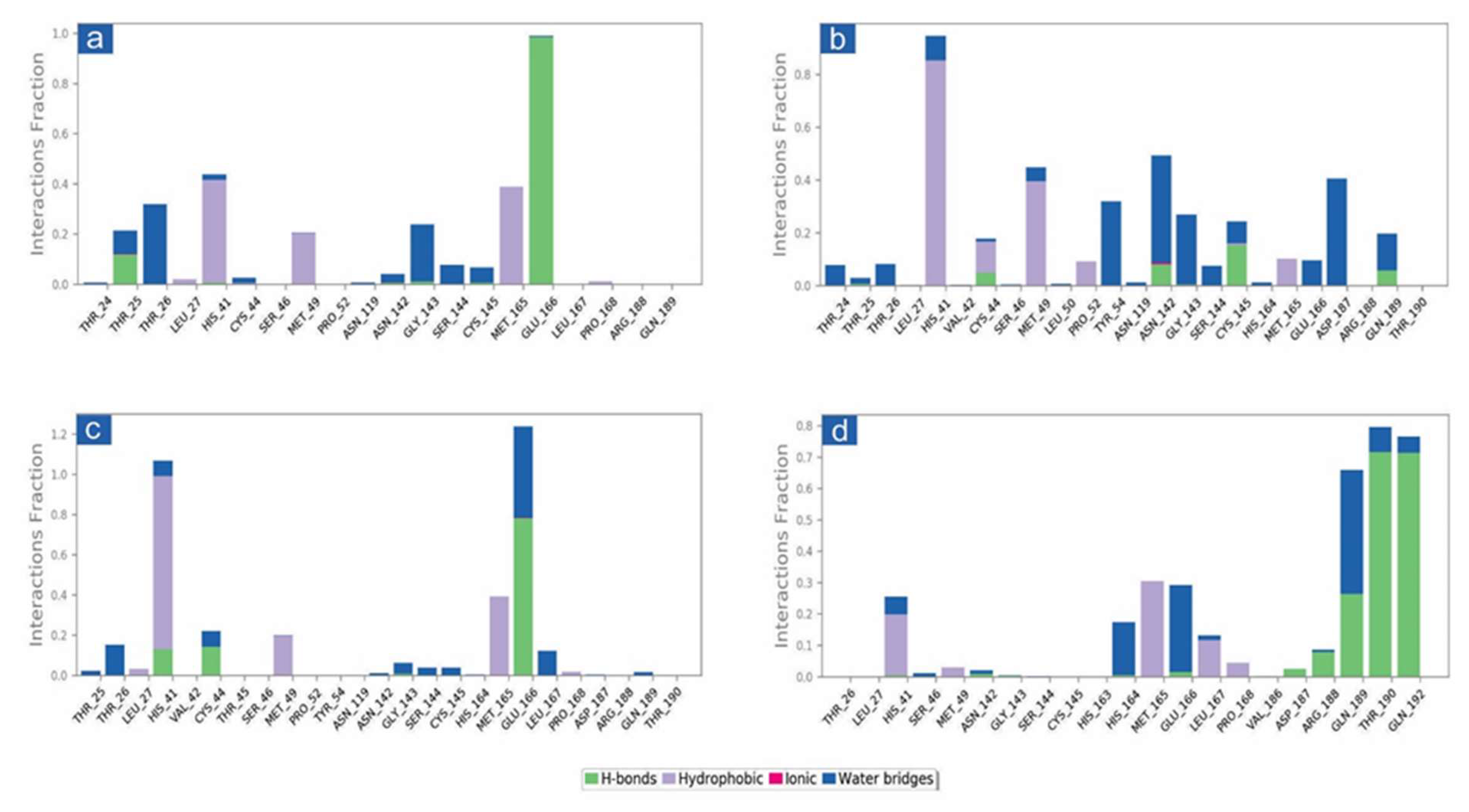
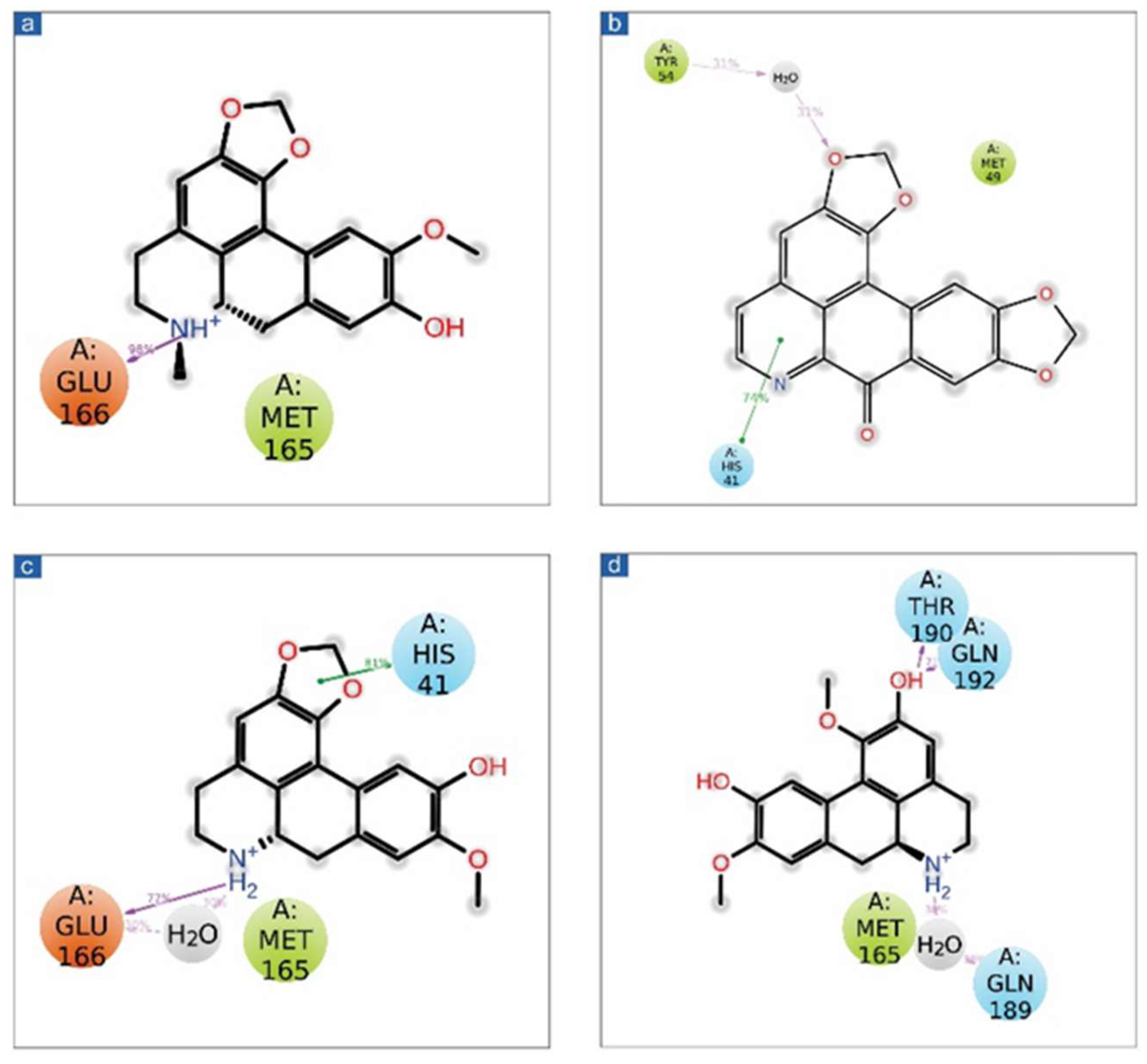
3.3.2. Protein-Ligand Interaction Profiling
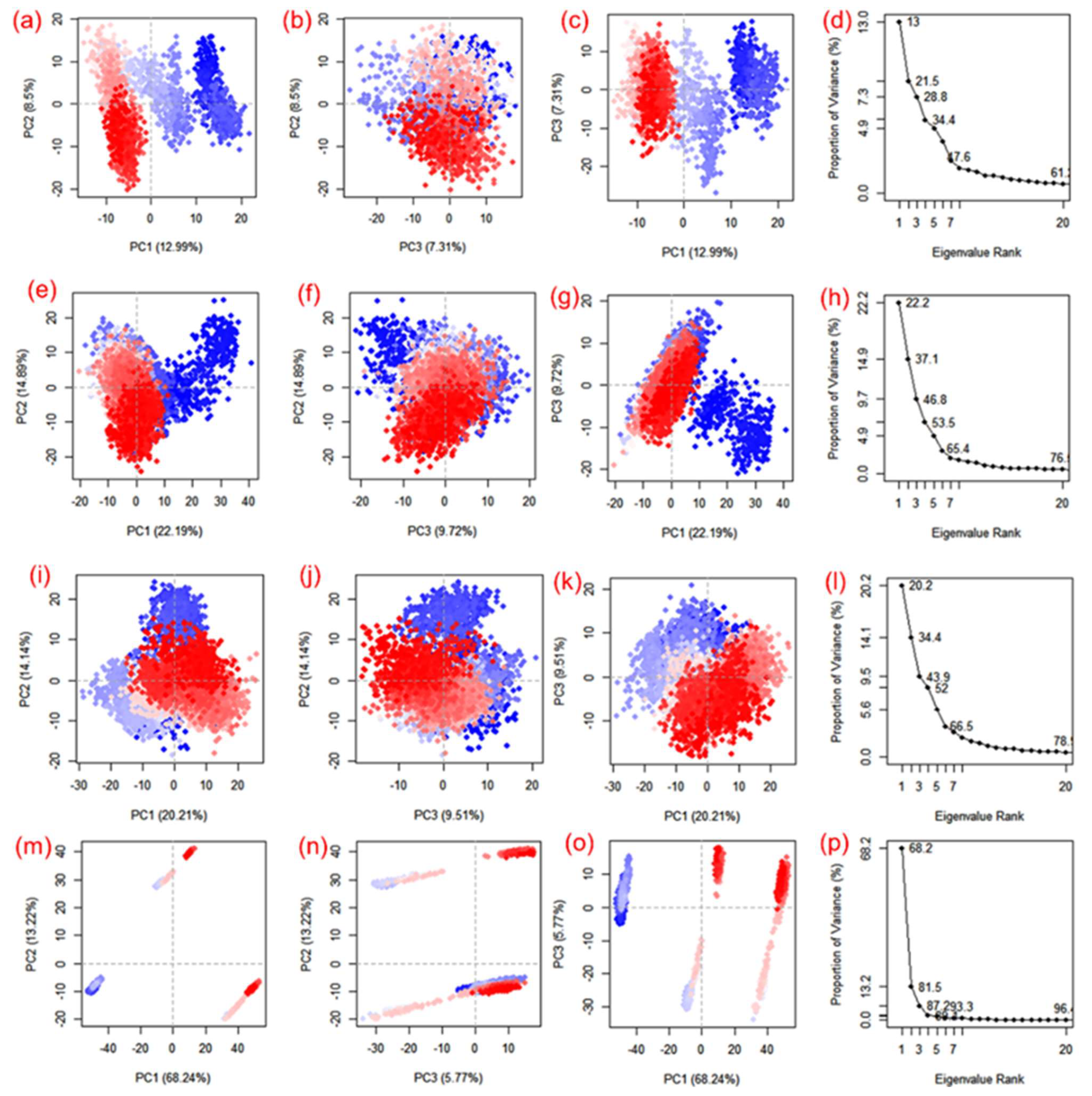
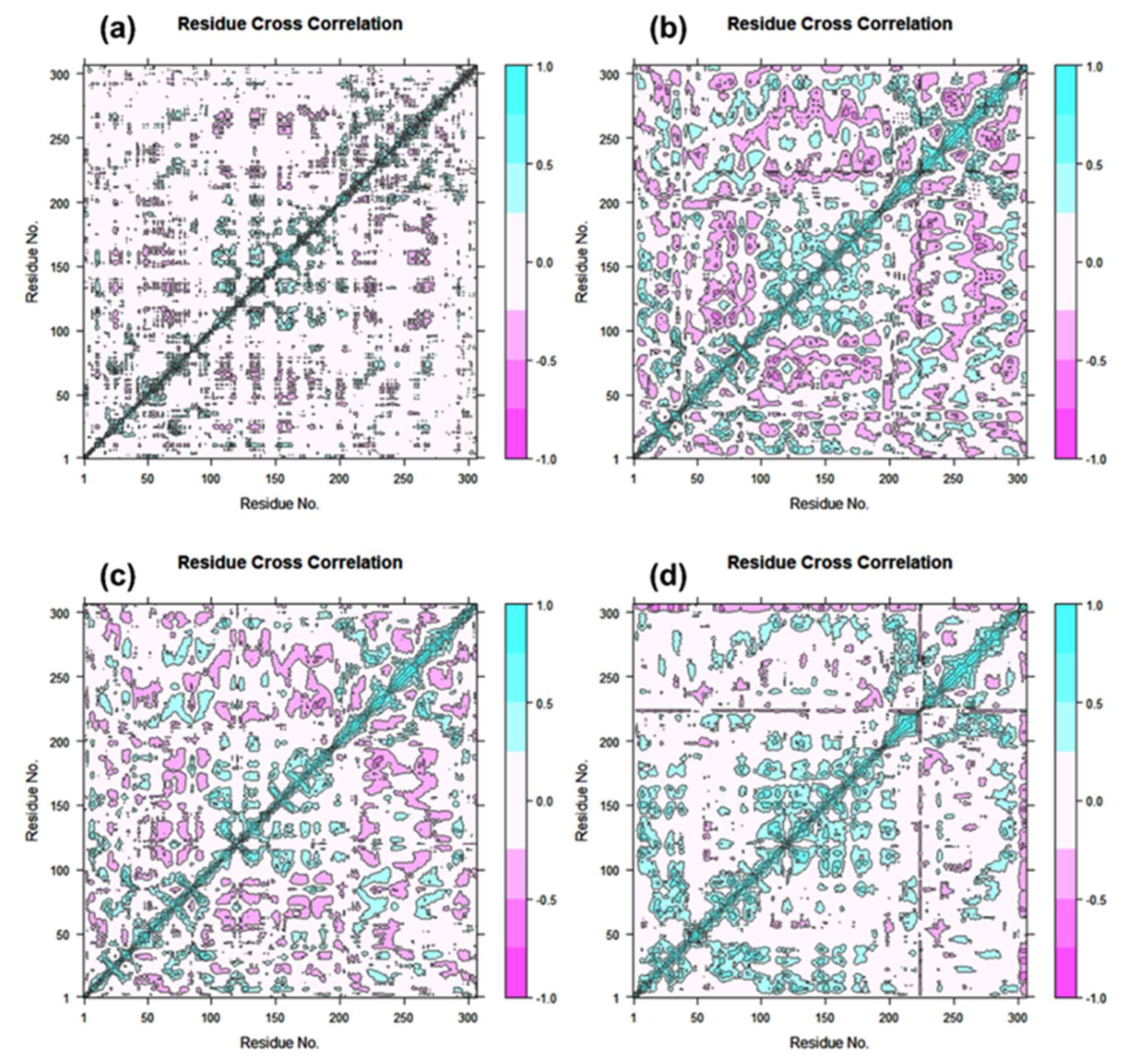
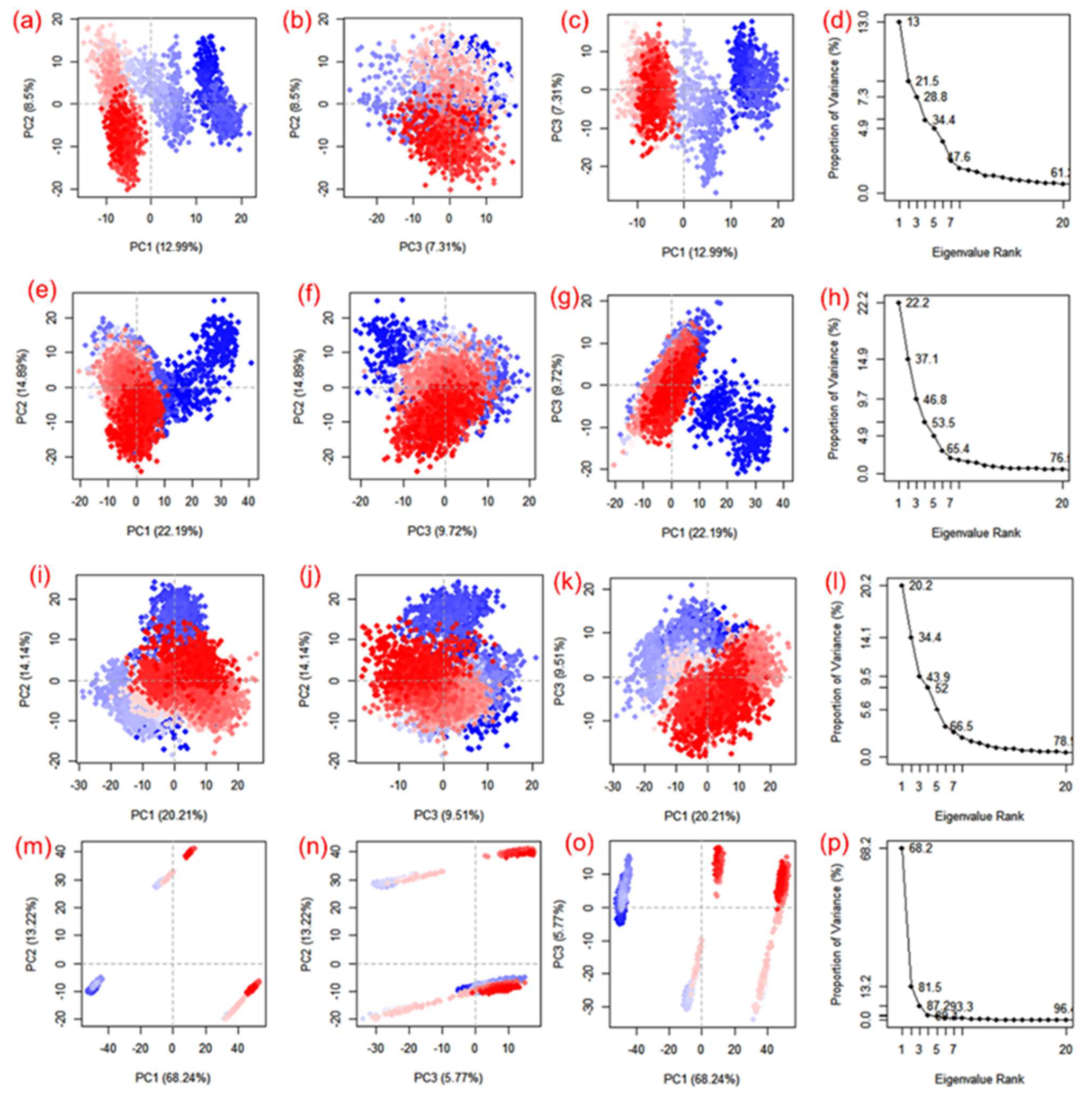
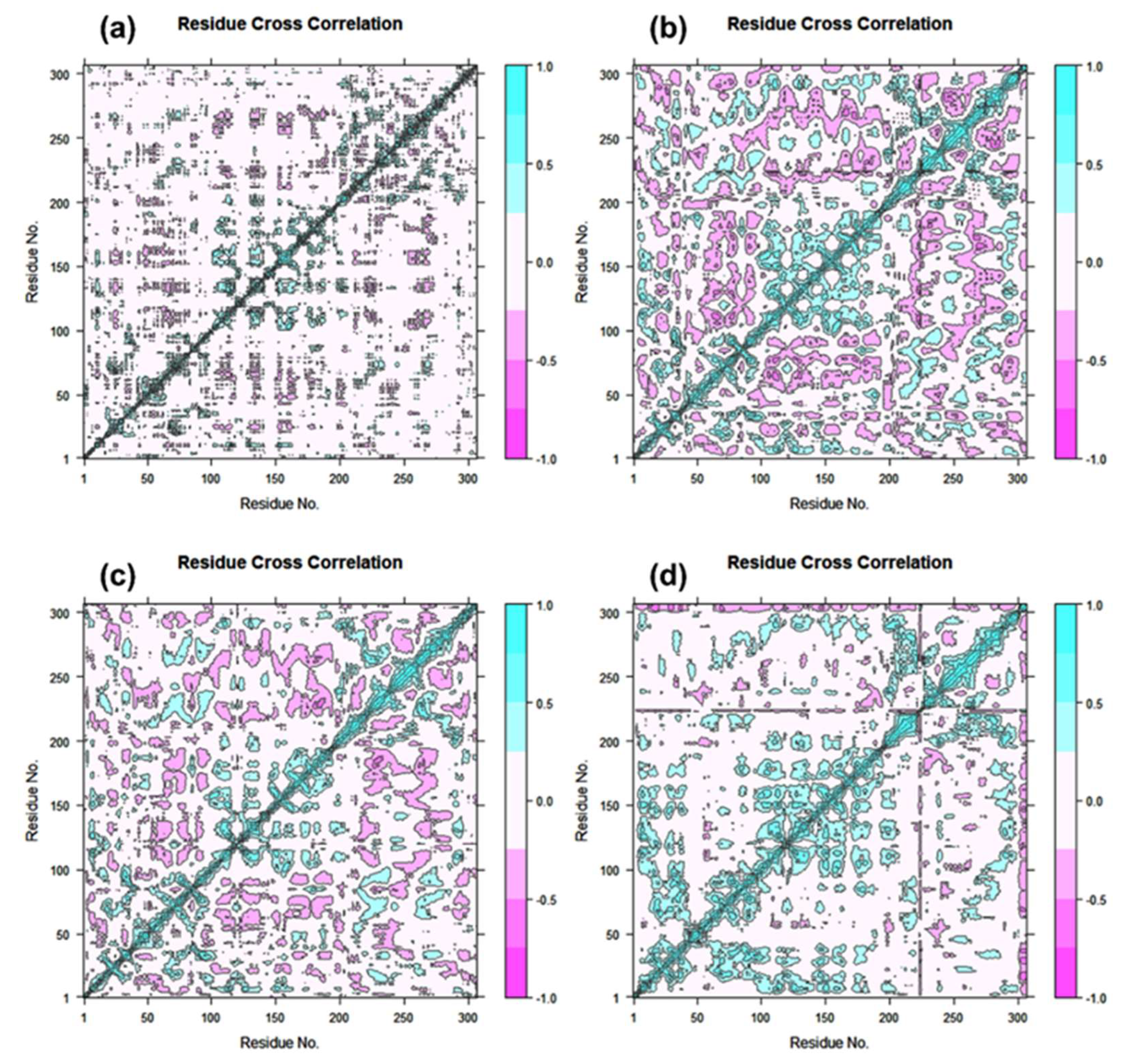
3.4. Essential Dynamics and Dynamic Cross-Correlation Matrix (DCCM) Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Al-Karmalawy, A.A.; Soltane, R.; Abo Elmaaty, A.; Tantawy, M.A.; Antar, S.A.; Yahya, G.; Chrouda, A.; Pashameah, R.A.; Mustafa, M.; Abu Mraheil, M.; et al. Coronavirus Disease (COVID-19) Control between Drug Repurposing and Vaccination: A Comprehensive Overview. Vaccines 2021, 9, 1317. [Google Scholar] [CrossRef]
- Farhat, N.; Khan, A.U. Repurposing Drug Molecule against SARS-Cov-2 (COVID-19) through Molecular Docking and Dynamics: A Quick Approach to Pick FDA-Approved Drugs. J. Mol. Model. 2021, 27, 312. [Google Scholar] [CrossRef]
- Elmorsy, M.A.; El-Baz, A.M.; Mohamed, N.H.; Almeer, R.; Abdel-Daim, M.M.; Yahya, G. In Silico Screening of Potent Inhibitors against COVID-19 Key Targets from a Library of FDA-Approved Drugs. Environ. Sci. Pollut. Res. 2022, 29, 12336–12346. [Google Scholar] [CrossRef] [PubMed]
- Jahan, I.; Onay, A. Potentials of Plant-Based Substance to Inhabit and Probable Cure for the COVID-19. Turk. J. Biol. 2020, 44, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.-L.; Jie-Ping, O.; Liu, Y.-C.; Hung, C.-P.; Tsai, M.-C.; Liao, P.-C.; Wang, E.I.-C.; Chen, Y.-L.; Su, Y.-C. Compositions and in Vitro Anticancer Activities of the Leaf and Fruit Oils of Litsea Cubeba from Taiwan. Nat. Prod. Commun. 2010, 5, 1934578X1000500425. [Google Scholar] [CrossRef] [Green Version]
- Si, L.; Chen, Y.; Han, X.; Zhan, Z.; Tian, S.; Cui, Q.; Wang, Y. Chemical Composition of Essential Oils of Litsea Cubeba Harvested from Its Distribution Areas in China. Molecules 2012, 17, 7057–7066. [Google Scholar] [CrossRef]
- Wong, M.H.; Lim, L.F.; bin Ahmad, F.; bin Assim, Z. Antioxidant and Antimicrobial Properties of Litsea Elliptica Blume and Litsea Resinosa Blume (Lauraceae). Asian Pac. J. Trop. Biomed. 2014, 4, 386–392. [Google Scholar] [CrossRef] [Green Version]
- Liao, P.-C.; Yang, T.-S.; Chou, J.-C.; Chen, J.; Lee, S.-C.; Kuo, Y.-H.; Ho, C.-L.; Chao, L.K.-P. Anti-Inflammatory Activity of Neral and Geranial Isolated from Fruits of Litsea Cubeba Lour. J. Funct. Foods 2015, 19, 248–258. [Google Scholar] [CrossRef]
- Kamle, M.; Mahato, D.K.; Lee, K.E.; Bajpai, V.K.; Gajurel, P.R.; Gu, K.S.; Kumar, P. Ethnopharmacological Properties and Medicinal Uses of Litsea Cubeba. Plants 2019, 8, 150. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Wang, C.F.; You, C.X.; Geng, Z.F.; Sun, R.Q.; Guo, S.S.; Du, S.S.; Liu, Z.L.; Deng, Z.W. Bioactivity of Essential Oil of Litsea Cubeba from China and Its Main Compounds against Two Stored Product Insects. J. Asia-Pac. Entomol. 2014, 17, 459–466. [Google Scholar] [CrossRef]
- Wang, H.; Liu, Y. Chemical Composition and Antibacterial Activity of Essential Oils from Different Parts of Litsea Cubeba. Chem. Biodivers. 2010, 7, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, Y.; Han, X.; Si, L.; Wu, Q.; Lin, L. Biology and Chemistry of Litsea Cubeba, a Promising Industrial Tree in China. J. Essent. Oil Res. 2013, 25, 103–111. [Google Scholar] [CrossRef]
- Agrawal, N.; Choudhary, A.S.; Sharma, M.C.; Dobhal, M.P. Chemical Constituents of Plants from the Genus Litsea. Chem. Biodivers. 2011, 8, 223–243. [Google Scholar] [CrossRef] [PubMed]
- Thielmann, J.; Muranyi, P. Review on the Chemical Composition of Litsea Cubeba Essential Oils and the Bioactivity of Its Major Constituents Citral and Limonene. J. Essent. Oil Res. 2019, 31, 361–378. [Google Scholar] [CrossRef]
- Cui, H.; Zhang, C.; Li, C.; Lin, L. Preparation and Antibacterial Activity of Litsea Cubeba Essential Oil/Dandelion Polysaccharide Nanofiber. Ind. Crops Prod. 2019, 140, 111739. [Google Scholar] [CrossRef]
- de Almeida, A.P.; Miranda, M.M.F.S.; Simoni, I.C.; Wigg, M.D.; Lagrota, M.H.C.; Costa, S.S. Flavonol Monoglycosides Isolated from the Antiviral Fractions of Persea Americana (Lauraceae) Leaf Infusion. Phytother. Res. 1998, 12, 562–567. [Google Scholar] [CrossRef]
- Tariq, S.; Wani, S.; Rasool, W.; Shafi, K.; Bhat, M.A.; Prabhakar, A.; Shalla, A.H.; Rather, M.A. A Comprehensive Review of the Antibacterial, Antifungal and Antiviral Potential of Essential Oils and Their Chemical Constituents against Drug-Resistant Microbial Pathogens. Microb. Pathog. 2019, 134, 103580. [Google Scholar] [CrossRef]
- Attia, Y.A.; Alagawany, M.M.; Farag, M.R.; Alkhatib, F.M.; Khafaga, A.F.; Abdel-Moneim, A.-M.E.; Asiry, K.A.; Mesalam, N.M.; Shafi, M.E.; Al-Harthi, M.A.; et al. Phytogenic Products and Phytochemicals as a Candidate Strategy to Improve Tolerance to Coronavirus. Front. Vet. Sci. 2020, 7, 573159. [Google Scholar] [CrossRef]
- Mishra, C.B.; Pandey, P.; Sharma, R.D.; Malik, M.Z.; Mongre, R.K.; Lynn, A.M.; Prasad, R.; Jeon, R.; Prakash, A. Identifying the Natural Polyphenol Catechin as a Multi-Targeted Agent against SARS-CoV-2 for the Plausible Therapy of COVID-19: An Integrated Computational Approach. Brief. Bioinform. 2021, 22, 1346–1360. [Google Scholar] [CrossRef]
- Pandey, P.; Rane, J.S.; Chatterjee, A.; Kumar, A.; Khan, R.; Prakash, A.; Ray, S. Targeting SARS-CoV-2 Spike Protein of COVID-19 with Naturally Occurring Phytochemicals: An in Silico Study for Drug Development. J. Biomol. Struct. Dyn. 2021, 39, 6306–6316. [Google Scholar] [CrossRef]
- Chikhale, R.V.; Sinha, S.K.; Patil, R.B.; Prasad, S.K.; Shakya, A.; Gurav, N.; Prasad, R.; Dhaswadikar, S.R.; Wanjari, M.; Gurav, S.S. In-Silico Investigation of Phytochemicals from Asparagus Racemosus as Plausible Antiviral Agent in COVID-19. J. Biomol. Struct. Dyn. 2021, 39, 5033–5047. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, M.K.; Singh, P.; Sharma, S.; Singh, T.P.; Ethayathulla, A.S.; Kaur, P. Identification of Bioactive Molecule from Withania Somnifera (Ashwagandha) as SARS-CoV-2 Main Protease Inhibitor. J. Biomol. Struct. Dyn. 2021, 39, 5668–5681. [Google Scholar] [CrossRef] [PubMed]
- Shree, P.; Mishra, P.; Selvaraj, C.; Singh, S.K.; Chaube, R.; Garg, N.; Tripathi, Y.B. Targeting COVID-19 (SARS-CoV-2) Main Protease through Active Phytochemicals of Ayurvedic Medicinal Plants—Withania Somnifera (Ashwagandha), Tinospora Cordifolia (Giloy) and Ocimum Sanctum (Tulsi)—A Molecular Docking Study. J. Biomol. Struct. Dyn. 2022, 40, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, P. In Silico Investigation of Phytoconstituents from Indian Medicinal Herb ‘Tinospora Cordifolia (Giloy)’ against SARS-CoV-2 (COVID-19) by Molecular Dynamics Approach. J. Biomol. Struct. Dyn. 2021, 39, 6792–6809. [Google Scholar] [CrossRef]
- Guan, Y.; Wang, D.; Tan, G.T.; Van Hung, N.; Cuong, N.M.; Pezzuto, J.M.; Fong, H.H.S.; Soejarto, D.D.; Zhang, H. Litsea Species as Potential Antiviral Plant Sources. Am. J. Chin. Med. 2016, 44, 275–290. [Google Scholar] [CrossRef] [Green Version]
- Yoon-Jae, S.; Gayong, P. Effects of Litsea Japonica Fruit Extract and Its Constituents on Hepatitis e Virus Replication. Rev. Clin. Pharmacol. Drug Ther. 2017, 15, 65. [Google Scholar]
- Mostafa, I.; Mohamed, N.H.; Mohamed, B.; Almeer, R.; Abulmeaty, M.M.A.; Bungau, S.G.; El-Shazly, A.M.; Yahya, G. In-Silico Screening of Naturally Derived Phytochemicals against SARS-CoV Main Protease. Environ. Sci. Pollut. Res. 2022, 29, 26775–26791. [Google Scholar] [CrossRef]
- Al Naggar, Y.; Giesy, J.P.; Abdel-Daim, M.M.; Javed Ansari, M.; Al-Kahtani, S.N.; Yahya, G. Fighting against the Second Wave of COVID-19: Can Honeybee Products Help Protect against the Pandemic? Saudi J. Biol. Sci. 2021, 28, 1519–1527. [Google Scholar] [CrossRef]
- Shaldam, M.A.; Yahya, G.; Mohamed, N.H.; Abdel-Daim, M.M.; Al Naggar, Y. In Silico Screening of Potent Bioactive Compounds from Honeybee Products against COVID-19 Target Enzymes. Environ. Sci. Pollut. Res. 2021, 28, 40507–40514. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and Discovery of Its Inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound Databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Labbé, C.M.; Rey, J.; Lagorce, D.; Vavruša, M.; Becot, J.; Sperandio, O.; Villoutreix, B.O.; Tufféry, P.; Miteva, M.A. MTiOpenScreen: A Web Server for Structure-Based Virtual Screening. Nucleic Acids Res. 2015, 43, W448–W454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Paul, P.; Yadav, P.; Kaul, R.; Maitra, S.S.; Jha, S.K.; Chaari, A. A Multi-Targeted Approach to Identify Potential Flavonoids against Three Targets in the SARS-CoV-2 Life Cycle. Comput. Biol. Med. 2022, 142, 105231. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Mohanty, U.; Noehre, J.; Sawyer, T.K.; Sherman, W.; Krilov, G. Probing the α-Helical Structural Stability of Stapled P53 Peptides: Molecular Dynamics Simulations and Analysis. Chem. Biol. Drug Des. 2010, 75, 348–359. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; Association for Computing Machinery: New York, NY, USA, 2006; p. 84-es. [Google Scholar]
- Grant, B.J.; Rodrigues, A.P.C.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S.D. Bio3d: An R Package for the Comparative Analysis of Protein Structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef] [Green Version]
- R Foundation for Statistical Computing. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013; Available online: http://www.R-project.org/2013 (accessed on 20 October 2021).
- Dwivedi, V.D.; Arya, A.; Yadav, P.; Kumar, R.; Kumar, V.; Raghava, G.P. DenvInD: Dengue Virus Inhibitors Database for Clinical and Molecular Research. Brief. Bioinform. 2021, 22, bbaa098. [Google Scholar] [CrossRef]
- Bharadwaj, S.; El-Kafrawy, S.A.; Alandijany, T.A.; Bajrai, L.H.; Shah, A.A.; Dubey, A.; Sahoo, A.K.; Yadava, U.; Kamal, M.A.; Azhar, E.I. Structure-Based Identification of Natural Products as SARS-CoV-2 Mpro Antagonist from Echinacea Angustifolia Using Computational Approaches. Viruses 2021, 13, 305. [Google Scholar] [CrossRef]
- Bharadwaj, S.; Lee, K.E.; Dwivedi, V.D.; Yadava, U.; Panwar, A.; Lucas, S.; Pandey, A.; Kang, S.G. Discovery of Ganoderma Lucidum Triterpenoids as Potential Inhibitors against Dengue Virus NS2B-NS3 Protease. Sci. Rep. 2019, 9, 19059. [Google Scholar] [CrossRef] [Green Version]
- Bharadwaj, S.; Lee, K.E.; Dwivedi, V.D.; Yadava, U.; Kang, S.G. Computational Aided Mechanistic Understanding of Camellia Sinensis Bioactive Compounds against Co-chaperone P23 as Potential Anticancer Agent. J. Cell. Biochem. 2019, 120, 19064–19075. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, S.; Lee, K.E.; Dwivedi, V.D.; Yadava, U.; Nees, M.; Kang, S.G. Density Functional Theory and Molecular Dynamics Simulation Support Ganoderma Lucidum Triterpenoids as Broad Range Antagonist of Matrix Metalloproteinases. J. Mol. Liq. 2020, 311, 113322. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
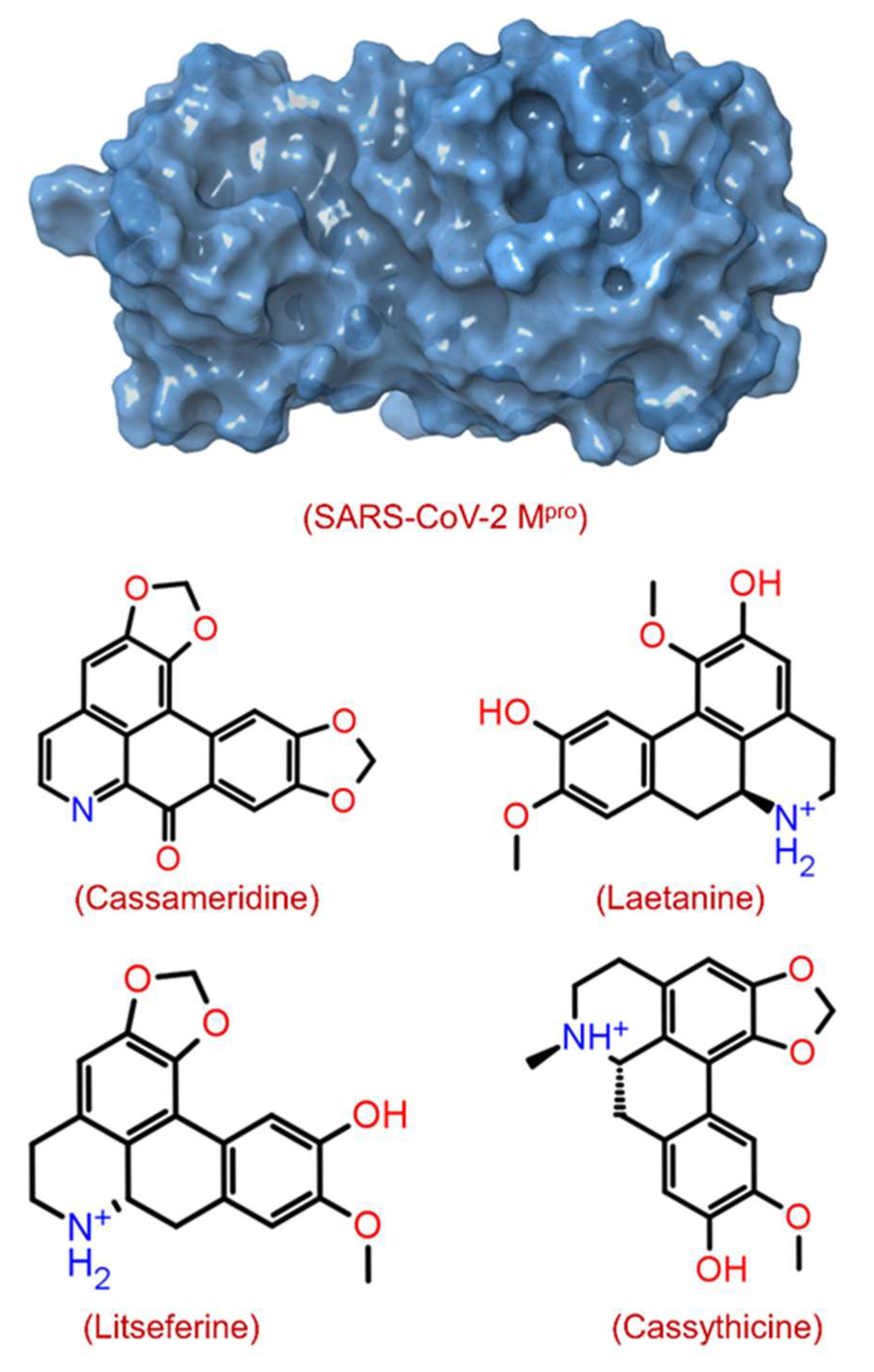
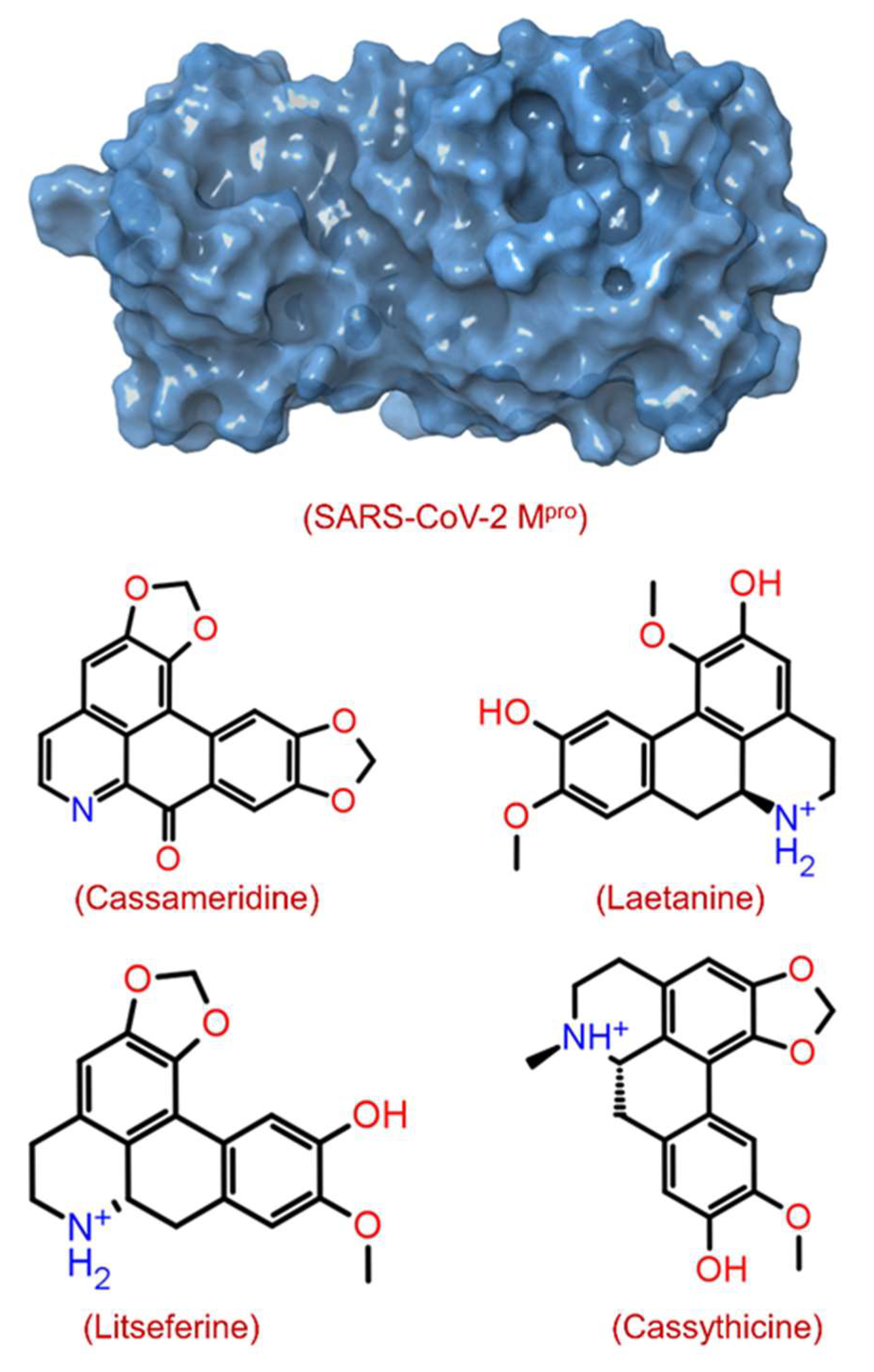
| S.no | Pubchem ID | Compound | Molecular wt. | Origin | Docking Score (kcal/mol) |
|---|---|---|---|---|---|
| 1 | 12302502 | Cassameridine | 319.3 | Cassytha filiformis | −9.3 |
| 2 | 129371873 | Laetanine | 313.3 | Ocotea teleiandra | −8.8 |
| 3 | 13891936 | Litseferine | 311.3 | Litsea glutinosa | −8.6 |
| 4 | 442194 | Cassythicine | 325.4 | Licaria sebifera | −8.6 |
| S.No. | Complex | H-Bond | Hydrophobic | Polar | π-π Stacking | Positive | Negative |
|---|---|---|---|---|---|---|---|
| 1 | SARS-CoV-2 Mpro-cassameridine | Gly143, Glu166 | Leu27, Met49, Tyr54, Cys145, Met165 | Thr25, Thr26, His41, Asn142, Gln189 | His41 | Arg188 | Glu166 Asp187 |
| 2 | SARS-CoV-2 Mpro-laetanine | Glu166 | Leu27, Met49, Tyr54, Leu141, Cys145, Met165 | Thr25, Thr26, His41, Asn142, Ser144, His163, His164 Gln189 | -- | Arg188 | Glu166 Asp187 |
| 3 | SARS-CoV-2 Mpro-litseferine | His41 Glu166 | Leu27, Met49, Tyr54, Cys145, Met165 | Thr25, Thr26, His41, Asn142, Ser144, His164 Gln189 | His41 | Arg188 | Glu166 Asp187 |
| 4 | SARS-CoV-2 Mpro-cassythicine | Gly143 Glu166 | Leu27, Val42, Met49, Tyr54, Cys145, Met165 | Thr25, His41, Asn142, His164 Gln189 | His41 | Arg188 | Glu166 Asp187 |
| 5 | SARS-CoV-2 Mpro-GC376 | Phe140 His163 Glu166 Gln189 | Met49, Tyr54, Phel40 Leu141 Cys145, Met165 Leu167 Pro168 Ala191 | His41, Asn142, Ser144, His163, His164 His172 Gln189 Thr190, Gln192 | -- | Arg188 | Glu166 Asp187 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bora, H.; Kamle, M.; Hassan, H.; Al-Emam, A.; Chopra, S.; Kirtipal, N.; Bharadwaj, S.; Kumar, P. Exploration of Potent Antiviral Phytomedicines from Lauraceae Family Plants against SARS-CoV-2 Main Protease. Viruses 2022, 14, 2783. https://doi.org/10.3390/v14122783
Bora H, Kamle M, Hassan H, Al-Emam A, Chopra S, Kirtipal N, Bharadwaj S, Kumar P. Exploration of Potent Antiviral Phytomedicines from Lauraceae Family Plants against SARS-CoV-2 Main Protease. Viruses. 2022; 14(12):2783. https://doi.org/10.3390/v14122783
Chicago/Turabian StyleBora, Himashree, Madhu Kamle, Hesham Hassan, Ahmed Al-Emam, Sidharth Chopra, Nikhil Kirtipal, Shiv Bharadwaj, and Pradeep Kumar. 2022. "Exploration of Potent Antiviral Phytomedicines from Lauraceae Family Plants against SARS-CoV-2 Main Protease" Viruses 14, no. 12: 2783. https://doi.org/10.3390/v14122783
APA StyleBora, H., Kamle, M., Hassan, H., Al-Emam, A., Chopra, S., Kirtipal, N., Bharadwaj, S., & Kumar, P. (2022). Exploration of Potent Antiviral Phytomedicines from Lauraceae Family Plants against SARS-CoV-2 Main Protease. Viruses, 14(12), 2783. https://doi.org/10.3390/v14122783