
Comparative Analysis of Tomato Brown Rugose Fruit Virus Isolates Shows Limited Genetic Diversity
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Real-Time RT-PCR Assays
2.3. Library Preparation and Sequencing
2.4. Bioinformatics Analyses
2.5. Phylogenetic Analysis
2.6. Estimation of Genetic Diversity Parameters and Neutrality Tests
2.7. Estimation of Gene Flow and Genetic Differentiation
2.8. Sequence Submission
3. Results
3.1. High-Throughput Sequencing Data
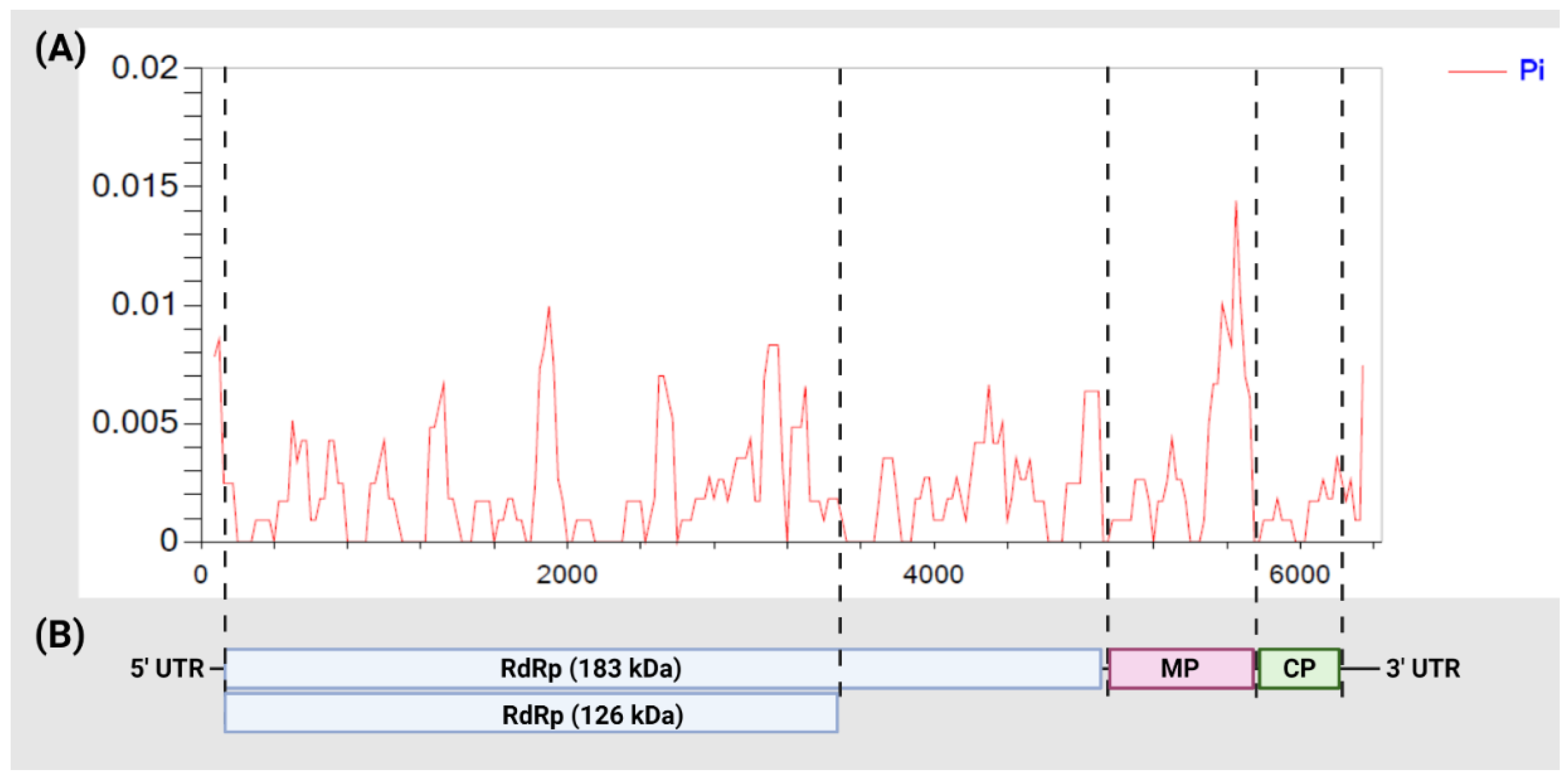
3.2. Diversity among ToBRFV Isolates
3.3. Mutations Affecting Movement Protein–Tm-22 Interaction
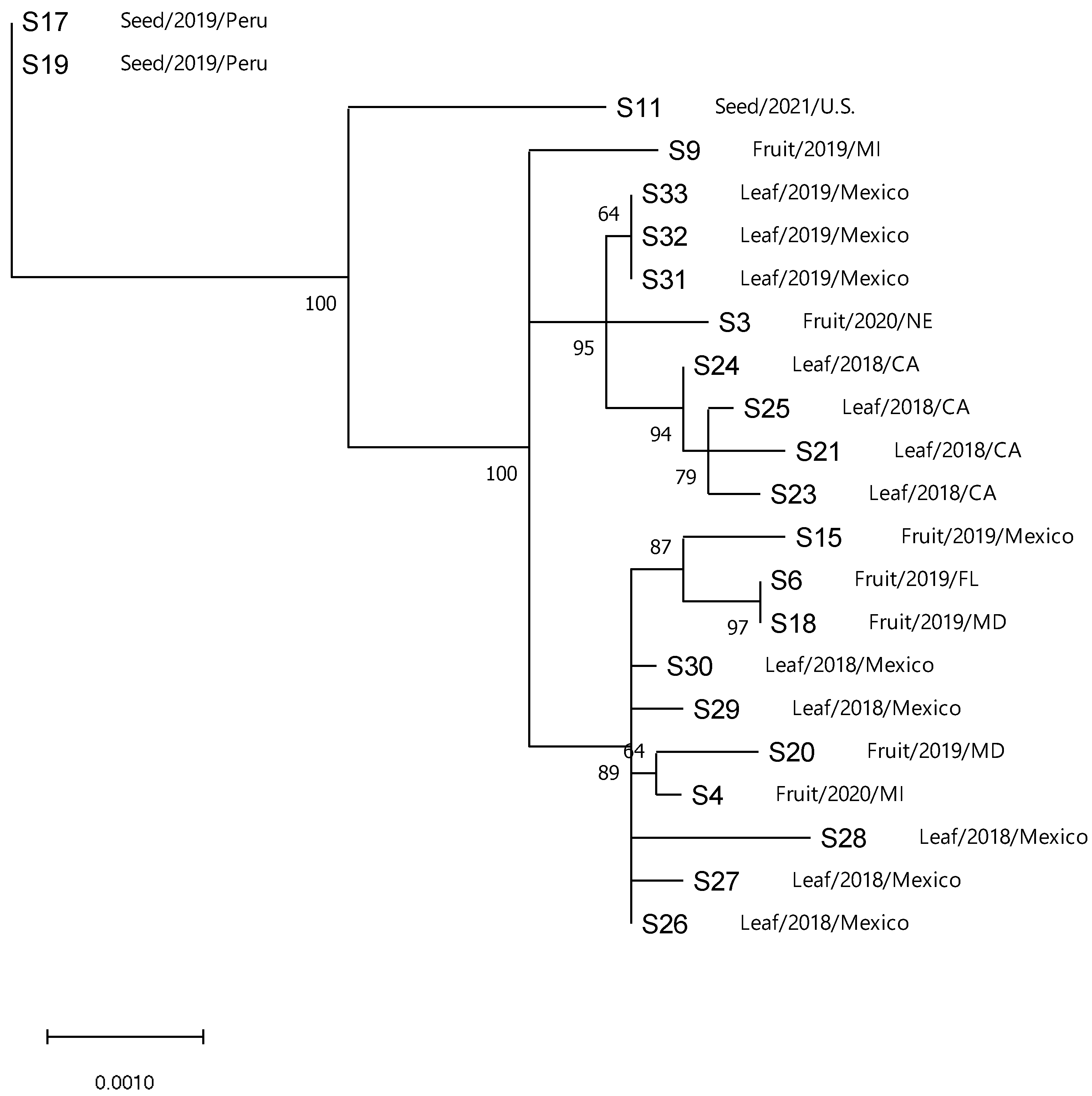
3.4. Phylogenetic Analysis of ToBRFV
3.5. Selection Pressure, Population Expansion
3.6. Gene Flow and Genetic Differentiation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Salem, N.; Mansour, A.; Ciuffo, M.; Falk, B.W.; Turina, M. A new tobamovirus infecting tomato crops in Jordan. Arch. Virol. 2016, 161, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Luria, N.; Smith, E.; Reingold, V.; Bekelman, I.; Lapidot, M.; Levin, I.; Elad, N.; Tam, Y.; Sela, N.; Abu-Ras, A.; et al. A New Israeli Tobamovirus Isolate Infects Tomato Plants Harboring Tm-22 Resistance Genes. PLoS ONE 2017, 12, e0170429. [Google Scholar] [CrossRef] [Green Version]
- Chanda, B.; Gilliard, A.; Jaiswal, N.; Ling, K.-S. Comparative Analysis of Host Range, Ability to Infect Tomato Cultivars with Tm-22 Gene, and Real-Time Reverse Transcription PCR Detection of Tomato Brown Rugose Fruit Virus. Plant Dis. 2021, 105, 3643–3652. [Google Scholar] [CrossRef] [PubMed]
- Davino, S.; Caruso, A.G.; Bertacca, S.; Barone, S.; Panno, S. Tomato brown rugose fruit virus: Seed Transmission Rate and Efficacy of Different Seed Disinfection Treatments. Plants 2020, 9, 1615. [Google Scholar] [CrossRef] [PubMed]
- Panno, S.; Caruso, A.G.; Barone, S.; Bosco, G.L.; Rangel, E.A.; Davino, S. Spread of Tomato Brown Rugose Fruit Virus in Sicily and Evaluation of the Spatiotemporal Dispersion in Experimental Conditions. Agronomy 2020, 10, 834. [Google Scholar] [CrossRef]
- Levitzky, N.; Smith, E.; Lachman, O.; Luria, N.; Mizrahi, Y.; Bakelman, H.; Sela, N.; Laskar, O.; Milrot, E.; Dombrovsky, A. The bumblebee Bombus terrestris carries a primary inoculum of Tomato brown rugose fruit virus contributing to disease spread in tomatoes. PLoS ONE 2019, 14, e0210871. [Google Scholar] [CrossRef] [PubMed]
- Salem, N.M.; Sulaiman, A.; Samarah, N.; Turina, M.; Vallino, M. Localization and Mechanical Transmission of Tomato Brown Rugose Fruit Virus in Tomato Seeds. Plant Dis. 2022, 106, 275–281. [Google Scholar] [CrossRef]
- Hak, H.; Spiegelman, Z. The Tomato Brown Rugose Fruit Virus Movement Protein Overcomes Tm-22 Resistance in Tomato While Attenuating Viral Transport. Mol. Plant-Microbe Interact. 2021, 34, 1024–1032. [Google Scholar] [CrossRef]
- Lanfermeijer, F.C.; Dijkhuis, J.; Sturre, M.J.; de Haan, P.; Hille, J. Cloning and characterization of the durable tomato mosaic virus resistance gene Tm-22 from Lycopersicon esculentum. Plant Mol. Biol. 2003, 52, 1039–1051. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Ma, H.; Wang, L.; Tettey, C.; Zhao, M.; Geng, C.; Tian, Y.; Li, X. Identification of genetic determinants of tomato brown rugose fruit virus that enable infection of plants harbouring the Tm-22 resistance gene. Mol. Plant Pathol. 2021, 22, 1347–1357. [Google Scholar] [CrossRef]
- Menzel, W.; Knierim, D.; Winter, S.; Hamacher, J.; Heupel, M. First report of Tomato brown rugose fruit virus infecting tomato in Germany. New Dis. Rep. 2019, 39, 1. [Google Scholar] [CrossRef] [Green Version]
- Fidan, H.; Sarikaya, P.; Calis, O. First report of Tomato brown rugose fruit virus on tomato in Turkey. New Dis. Rep. 2019, 39, 18. [Google Scholar] [CrossRef] [Green Version]
- Panno, S.; Caruso, A.G.; Davino, S. First Report of Tomato Brown Rugose Fruit Virus on Tomato Crops in Italy. Plant Dis. 2019, 103, 1443. [Google Scholar] [CrossRef]
- Ling, K.-S.; Tian, T.; Gurung, S.; Salati, R.; Gilliard, A. First Report of Tomato Brown Rugose Fruit Virus Infecting Greenhouse Tomato in the United States. Plant Dis. 2019, 103, 1439. [Google Scholar] [CrossRef]
- Dey, K.; Velez-Climent, M.; Soria, P.; Batuman, O.; Mavrodieva, V.; Wei, G.; Zhou, J.; Adkins, S.; McVay, J. First report of Tomato brown rugose fruit virus infecting tomato in Florida, USA. New Dis. Rep. 2021, 44, e12028. [Google Scholar] [CrossRef]
- Skelton, A.; Buxton-Kirk, A.; Ward, R.; Harju, V.; Frew, L.; Fowkes, A.; Long, M.; Negus, A.; Forde, S.; Adams, I.; et al. First report of Tomato brown rugose fruit virus in tomato in the United Kingdom. New Dis. Rep. 2019, 40, 12. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.-Y.; Ma, H.-Y.; Han, S.-L.; Geng, C.; Tian, Y.-P.; Li, X.-D. First Report of Tomato brown rugose fruit virus Infecting Tomato in China. Plant Dis. 2019, 103, 2973. [Google Scholar] [CrossRef]
- Alkowni, R.; Alabdallah, O.; Fadda, Z. Molecular identification of tomato brown rugose fruit virus in tomato in Palestine. J. Plant Pathol. 2019, 101, 719–723. [Google Scholar] [CrossRef]
- Davidson, K. Tomato Brown Rugose Fruit Virus Identified in Ontario. 2019. Available online: http://thegrower.org/news/tomato-brown-rugose-fruit-virus-identified-ontario (accessed on 4 January 2021).
- Camacho-Beltrán, E.; Pérez-Villarreal, A.; Leyva-López, N.E.; Rodríguez-Negrete, E.A.; Ceniceros-Ojeda, E.A.; Méndez-Lozano, J. Occurrence of Tomato brown rugose fruit virus Infecting Tomato Crops in Mexico. Plant Dis. 2019, 103, 1440. [Google Scholar] [CrossRef]
- Beris, D.; Malandraki, I.; Kektsidou, O.; Theologidis, I.; Vassilakos, N.; Varveri, C. First Report of Tomato Brown Rugose Fruit Virus Infecting Tomato in Greece. Plant Dis. 2020, 104, 2035. [Google Scholar] [CrossRef]
- Amer, M.A.; Mahmoud, S.Y. First report of Tomato brown rugose fruit virus on tomato in Egypt. New Dis. Rep. 2020, 41, 24. [Google Scholar] [CrossRef]
- Alfaro-Fernández, A.; Castillo, P.; Sanahuja, E.; Rodríguez-Salido, M.C.; Font, M.I. First Report of Tomato Brown Rugose Fruit Virus in Tomato in Spain. Plant Dis. 2021, 105, 515. [Google Scholar] [CrossRef]
- van de Vossenberg, B.T.L.H.; Visser, M.; Bruinsma, M.; Koenraadt, H.M.S.; Westenberg, M.; Botermans, M. Real-time tracking of Tomato brown rugose fruit virus (ToBRFV) outbreaks in the Netherlands using Nextstrain. PLoS ONE 2020, 15, e0234671. [Google Scholar] [CrossRef]
- EPPO. Tomato Brown Rugose Fruit Virus. EPPO Datasheets on Pests Recommended for Regulation. 2021. Available online: https://gd.eppo.int (accessed on 20 May 2022).
- Hasan, Z.M.; Salem, N.M.; Ismail, I.D.; Akel, E.H.; Ahmad, A.Y. First Report of Tomato Brown Rugose Fruit Virus on Greenhouse Tomato in Syria. Plant Dis. 2022, 106, 772. [Google Scholar] [CrossRef]
- Ghorbani, A.; Rostami, M.; Seifi, S.; Izadpanah, K. First report of Tomato brown rugose fruit virus in greenhouse tomato in Iran. New Dis. Rep. 2021, 44, e12040. [Google Scholar] [CrossRef]
- Sabra, A.; Al-Saleh, M.A.; Al-Shahwan, I.M.; Amer, M.A. First Report of Tomato Brown Rugose Fruit Virus Infecting the Tomato Crop in Saudi Arabia. Plant Dis. 2022, 106, 1310. [Google Scholar] [CrossRef] [PubMed]
- van de Vossenberg, B.T.L.H.; Dawood, T.; Woźny, M.; Botermans, M. First Expansion of the Public Tomato Brown Rugose Fruit Virus (ToBRFV) Nextstrain Build; Inclusion of New Genomic and Epidemiological Data. Phytofrontiers 2021, 1, 359–363. [Google Scholar] [CrossRef]
- Maayan, Y.; Pandaranayaka, E.P.J.; Srivastava, D.A.; Lapidot, M.; Levin, I.; Dombrovsky, A.; Harel, A. Using genomic analysis to identify tomato Tm-2 resistance-breaking mutations and their underlying evolutionary path in a new and emerging tobamovirus. Arch. Virol. 2018, 163, 1863–1875. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Antipov, D.; Raiko, M.; Lapidus, A.; Pevzner, P.A. Metaviral SPAdes: Assembly of viruses from metagenomic data. Bioinformatics 2020, 36, 4126–4129. [Google Scholar] [CrossRef]
- Lu, J.-J.; He, E.-Q.; Bao, W.-Q.; Chen, J.-S.; Sun, S.-R.; Gao, S.-J. Comparative genomics reveals insights into genetic variability and molecular evolution among sugarcane yellow leaf virus populations. Sci. Rep. 2021, 11, 7149. [Google Scholar] [CrossRef]
- Velazquez-Salinas, L.; Zarate, S.; Eberl, S.; Gladue, D.P.; Novella, I.; Borca, M.V. Positive Selection of ORF1ab, ORF3a, and ORF8 Genes Drives the Early Evolutionary Trends of SARS-CoV-2 During the 2020 COVID-19 Pandemic. Front. Microbiol. 2020, 11, 550674. [Google Scholar] [CrossRef]
- Chanda, B.; Rivera, Y.; Nunziata, S.O.; Galvez, M.E.; Gilliard, A.; Ling, K.-S. Complete Genome Sequence of a Tomato Brown Rugose Fruit Virus Isolated in the United States. Genome Announc. 2020, 9, e00630-20. [Google Scholar] [CrossRef]
- Rivarez, M.P.S.; Vučurović, A.; Mehle, N.; Ravnikar, M.; Kutnjak, D. Global Advances in Tomato Virome Research: Current Status and the Impact of High-Throughput Sequencing. Front. Microbiol. 2021, 12, 671925. [Google Scholar] [CrossRef]
- Taraporewala, Z.F.; Culver, J.N. Structural and Functional Conservation of the Tobamovirus Coat Protein Elicitor Active Site. Mol. Plant-Microbe Interact. 1997, 10, 597–604. [Google Scholar] [CrossRef] [Green Version]
- Fraile, A.; Escriu, F.; Aranda, M.A.; Malpica, J.M.; Gibbs, A.J.; García-Arenal, F. A century of tobamovirus evolution in an Australian population of Nicotiana glauca. J. Virol. 1997, 71, 8316–8320. [Google Scholar] [CrossRef] [Green Version]
- Fraile, A.; Malpica, J.M.; Aranda, M.A.; Rodriguez-Cerezo, E.; García-Arenal, F.; Rodriguez, F.G.-A. Genetic Diversity in Tobacco Mild Green Mosaic Tobamovirus Infecting the Wild PlantNicotiana glauca. Virology 1996, 223, 148–155. [Google Scholar] [CrossRef] [Green Version]
- Fraile, A.; Pagán, I.; Anastasio, G.; Sáez, E.; García-Arenal, F. Rapid genetic diversification and high fitness penalties associated with pathogenicity evolution in a plant virus. Mol. Biol. Evol. 2011, 28, 1425–1437. [Google Scholar] [CrossRef]
- Janzac, B.; Fabre, F.; Palloix, A.; Moury, B. Constraints on evolution of virus avirulence factors predict the durability of corresponding plant resistances. Mol. Plant Pathol. 2009, 10, 599–610. [Google Scholar] [CrossRef]
- Branthôme, F.-X. ToBRFV: Latest Update. Available online: http://www.tomatonews.com/en/tobrfv-latest-update_2_997.html (accessed on 30 June 2021).
- HortiDaily. 2020. Available online: https://www.hortidaily.com/article/9189118/netherlands-peruvian-tomato-seed-with-tobrfv-intercepted/ (accessed on 20 July 2021).


{kind=link}
{kind=link}
{kind=link}
| Isolate | Plant Material | Date Received | Source | Location a | Real-Time PCR (Ct) b |
|---|---|---|---|---|---|
| S3 | Fruit | April 2020 | Retail store | Nebraska, U.S. | 7.9 |
| S4 | Fruit | February 2020 | Retail store | Michigan, U.S. | 6.7 |
| S6 | Fruit | November 2019 | Retail store | Florida, U.S. | 9.4 |
| S9 | Fruit | December 2019 | Retail store | Michigan, U.S. | 11.1 |
| S11 | Seed | March 2021 | Seed producer | U.S. c | 9.8 |
| S15 | Fruit | December 2019 | Import | Mexico | 6.5 |
| S17 | Seed | August 2019 | Seed producer | Peru | 16.5 |
| S18 | Fruit | December 2019 | Retail store | Maryland, U.S. | NT d |
| S19 | Seed | August 2019 | Seed producer | Peru | 9.5 |
| S20 | Fruit | December 2019 | Retail store | Maryland, U.S. | 8.7 |
| S21 | Leaf | November 2018 | Greenhouse | California, U.S. | NT |
| S23 | Leaf | November 2018 | Greenhouse | California, U.S. | 24.82 |
| S24 | Leaf | November 2018 | Greenhouse | California, U.S. | 21.12 |
| S25 | Leaf | November 2018 | Greenhouse | California, U.S. | NT |
| S26 | Leaf | November 2018 | Greenhouse | Mexico | 6.76 |
| S27 | Leaf | November 2018 | Greenhouse | Mexico | 8.26 |
| S28 | Leaf | November 2018 | Greenhouse | Mexico | 6.35 |
| S29 | Leaf | November 2018 | Greenhouse | Mexico | 6.26 |
| S30 | Leaf | November 2018 | Greenhouse | Mexico | 16.38 |
| S31 | Leaf | February 2019 | Greenhouse | Mexico | 14.02 |
| S32 | Leaf | February 2019 | Greenhouse | Mexico | 11.91 |
| S33 | Leaf | February 2019 | Greenhouse | Mexico | 8.14 |
| Isolate | Raw Reads | No. Mapped Reads a | Mean Coverage | % RefSeq b | Draft Genome Size (bp) |
|---|---|---|---|---|---|
| S3 | 13,462,663 | 3,809,241 | 42,288 | 99.8 | 6376 |
| S4 | 14,057,467 | 9,734,997 | 117,165 | 99.9 | 6380 |
| S6 | 15,273,550 | 11,035,573 | 125,255 | 99.9 | 6380 |
| S9 | 13.042,460 | 2,771,812 | 31,097 | 99.9 | 6380 |
| S11 | 50,476,803 | 2,386,377 | 26,954 | 99.9 | 6382 |
| S15 | 12,246,915 | 8,521,243 | 96,252 | 99.9 | 6381 |
| S17 | 66,390,808 | 3,177,034 | 35,528 | 99.9 | 6381 |
| S18 | 63,394,513 | 51,801,176 | 715,817 | 99.9 | 6392 |
| S19 | 78,158,737 | 65,509,729 | 773,145 | 99.9 | 6393 |
| S20 | 226,676,066 | 48,291,439 | 604,169 | 99.6 | 6375 |
| S21 | 47,616,520 | 46,470,185 | 598,411 | 99.9 | 6384 |
| S23 | 32,736,111 | 6701 | 78 | 99.4 | 6357 |
| S24 | 33,270,742 | 8448 | 99 | 99.6 | 6371 |
| S25 | 20,186,935 | 8,722,599 | 104,912 | 99.9 | 6392 |
| S26 | 26,239,529 | 23,870,654 | 313,051 | 99.9 | 6384 |
| S27 | 35,553,785 | 16,118,484 | 202,182 | 99.9 | 6380 |
| S28 | 19,230,903 | 17,252,098 | 218,573 | 99.8 | 6383 |
| S29 | 12,671,194 | 8,202,718 | 101,900 | 99.8 | 6383 |
| S30 | 39,047,819 | 14,943,377 | 184,625 | 100.0 | 6393 |
| S31 | 35,645,442 | 31,696,564 | 408,411 | 99.9 | 6384 |
| S32 | 48,390,993 | 35,607,924 | 448,364 | 99.9 | 6386 |
| S33 | 30,027,166 | 26,643,010 | 342,051 | 99.8 | 6384 |
| Gene | Sequence Length (bp) | Nt Identity ID % a | A.A. Identity % b | Synonymous Mutations | Non-Synonymous Mutation |
|---|---|---|---|---|---|
| 183 kDa | 4848 | 99.78 | 99.94 | 53 | 8 |
| 126 kDa | 3351 | 99.78 | 99.96 | 36 | 4 |
| MP | 801 | 99.63 | 99.41 | 10 | 6 |
| CP | 480 | 99.88 | 99.71 | 1 | 4 |
| Gene | Sequence Length (bp) | N a | H b | Sc | π d | Hd e | Tajima’s D f | Fu and Li’s D * g | Fu and Li’s F * h | dN/dS i |
|---|---|---|---|---|---|---|---|---|---|---|
| RdRp (183 kDa) | 4848 | 22 | 17 | 60 | 0.00220 | 0.974 | −1.44271 | −1.55880 | −1.78233 | 0.029 |
| 144 j | 85 | 220 | 0.00238 | 0.987 | −2.32187 ** | −3.83944 ** | −3.75501 ** | 0.094 | ||
| RdRp (126 kDa) | 3351 | 22 | 16 | 40 | 0.00219 | 0.965 | −1.29515 | −1.41931 | −1.61486 | 0.019 |
| 144 j | 78 | 149 | 0.00221 | 0.981 | −2.34105 ** | −3.84386 ** | −3.78484 ** | 0.079 | ||
| MP | 801 | 22 | 11 | 17 | 0.00362 | 0.892 | −1.38241 | −1.30950 | −1.55103 | 0.377 |
| 145 | 40 | 48 | 0.00414 | 0.923 | −1.93509 * | −3.47504 ** | −3.38850 ** | 0.192 | ||
| CP | 480 | 22 | 6 | 5 | 0.00112 | 0.476 | −1.80901 * | −2.09702 | −2.33018 | 1.525 |
| 145 | 22 | 25 | 0.00190 | 0.644 | −2.27816 ** | −3.92019 ** | −3.92314 ** | 0.116 | ||
| Full-length | 6117 | 22 | 18 | 82 | 0.00230 | 0.978 | −1.52835 | −1.66511 | −1.90015 | 0.123 k |
| 145 | 96 | 294 | 0.00257 | 0.992 | −2.31476 ** | −4.15842 ** | −3.94355 ** | 0.413 k |
| Population | Fst a | Nm b | Kst * | Z * | Snn |
|---|---|---|---|---|---|
| Clade 1 vs. 2 | 0.42279 | 0.34 | 0.21000 * c | 1.67244 * | 1.0 nsd |
| Clade 1 vs. 3 | 0.53847 | 0.21 | 0.02486 *** | 8.14475 *** | 1.0 *** |
| Clade 2 vs. 3 | 0.36199 | 0.44 | 0.00671 *** | 8.16035 *** | 1.0 *** |
| Clade 1 and 2 vs. 3 | 0.40248 | 0.37 | 0.02507 *** | 8.16805 *** | 1.0 *** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abrahamian, P.; Cai, W.; Nunziata, S.O.; Ling, K.-S.; Jaiswal, N.; Mavrodieva, V.A.; Rivera, Y.; Nakhla, M.K. Comparative Analysis of Tomato Brown Rugose Fruit Virus Isolates Shows Limited Genetic Diversity. Viruses 2022, 14, 2816. https://doi.org/10.3390/v14122816
Abrahamian P, Cai W, Nunziata SO, Ling K-S, Jaiswal N, Mavrodieva VA, Rivera Y, Nakhla MK. Comparative Analysis of Tomato Brown Rugose Fruit Virus Isolates Shows Limited Genetic Diversity. Viruses. 2022; 14(12):2816. https://doi.org/10.3390/v14122816
Chicago/Turabian StyleAbrahamian, Peter, Weili Cai, Schyler O. Nunziata, Kai-Shu Ling, Namrata Jaiswal, Vessela A. Mavrodieva, Yazmín Rivera, and Mark K. Nakhla. 2022. "Comparative Analysis of Tomato Brown Rugose Fruit Virus Isolates Shows Limited Genetic Diversity" Viruses 14, no. 12: 2816. https://doi.org/10.3390/v14122816
APA StyleAbrahamian, P., Cai, W., Nunziata, S. O., Ling, K.-S., Jaiswal, N., Mavrodieva, V. A., Rivera, Y., & Nakhla, M. K. (2022). Comparative Analysis of Tomato Brown Rugose Fruit Virus Isolates Shows Limited Genetic Diversity. Viruses, 14(12), 2816. https://doi.org/10.3390/v14122816