
Structure Based Affinity Maturation and Characterizing of SARS-CoV Antibody CR3022 against SARS-CoV-2 by Computational and Experimental Approaches



Abstract
:1. Introduction
2. Results and Discussion
2.1. Computational Virtual Mutation
2.2. Binding of the Wild-Type and Variants of CR3022 to SARS-CoV-2 RBD and SARS-CoV RBD
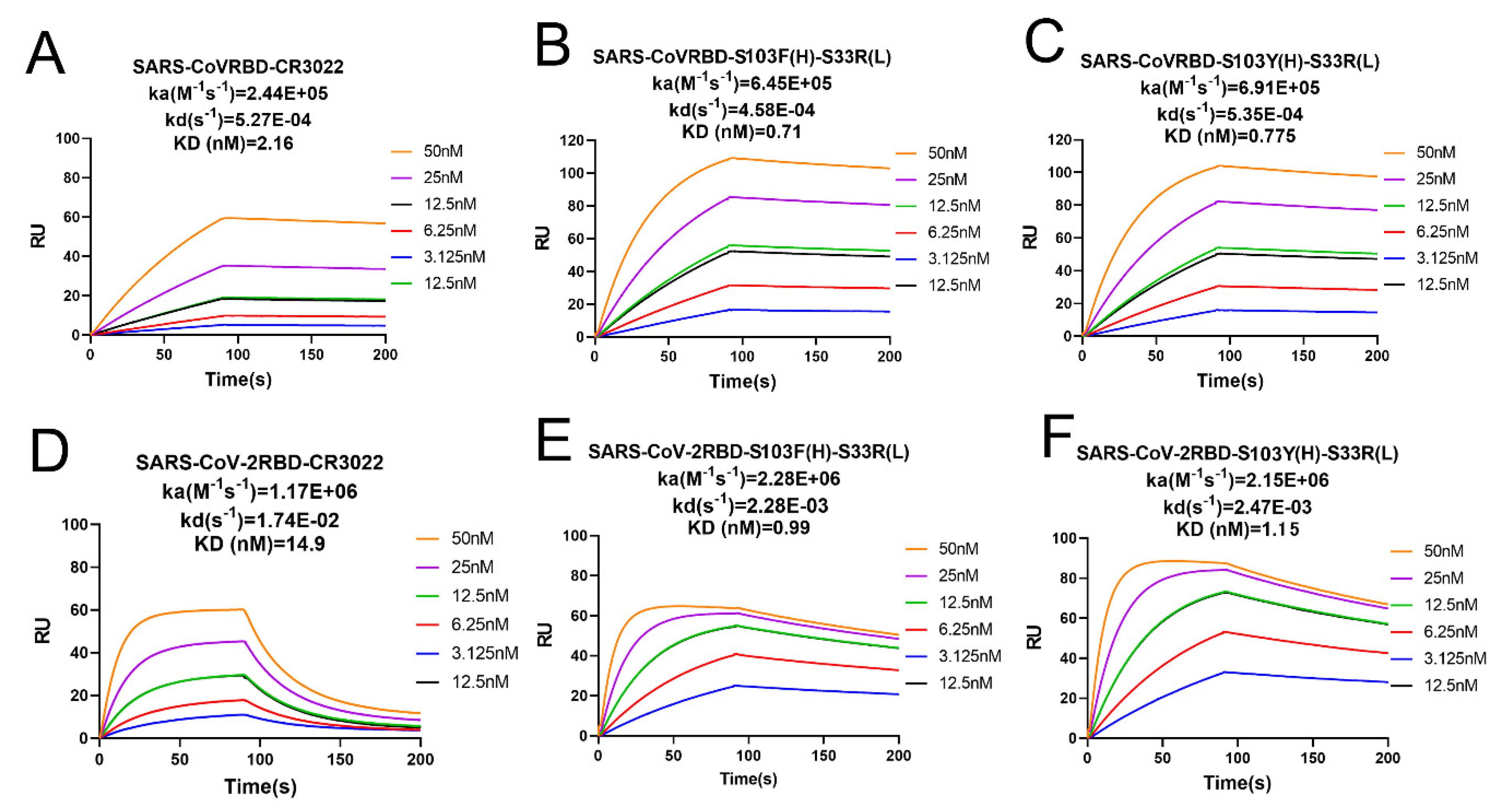
2.3. Binding Affinity Determined by SPR Analysis
2.4. Auto-Reactivity of the Wild-Type and Variants of CR3022
2.5. Structural Stability of Two Double-Site Mutant Antibodies
2.6. Binding Free Energy Calculation of Wild-Type CR3022 and Two Mutated Antibodies
2.7. Per-Residue Energy Decomposition
2.8. Distance Analysis of Key Secondary Structures
2.9. The Binding Profiles Analysis
3. Materials and Methods
3.1. Structure Preparation
3.2. Virtual Mutation
3.3. Protein Expression and Purification
3.4. Antibody Binding Detection by ELISA
3.5. Antibody Affinity Detection by SPR
3.6. Auto-Immune Reactivity Test
3.7. Molecular Dynamics (MD) Simulation
3.8. Binding Free Energy Calculation by MM/PBSA
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van Dorp, L.; Acman, M.; Richard, D.; Shaw, L.P.; Ford, C.E.; Ormond, L.; Owen, C.J.; Pang, J.; Tan, C.C.; Boshier, F.A. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect. Genet. Evol. 2020, 83, 104351. [Google Scholar] [CrossRef]
- Vandelli, A.; Monti, M.; Milanetti, E.; Armaos, A.; Rupert, J.; Zacco, E.; Bechara, E.; Delli Ponti, R.; Tartaglia, G.G. Structural analysis of SARS-CoV-2 genome and predictions of the human interactome. Nucleic Acids Res. 2020, 48, 11270–11283. [Google Scholar] [CrossRef]
- Chen, J.; Wang, R.; Wang, M.; Wei, G.-W. Mutations strengthened SARS-CoV-2 infectivity. J. Mol. Biol. 2020, 432, 5212–5226. [Google Scholar] [CrossRef] [PubMed]
- Riva, L.; Yuan, S.; Yin, X.; Martin-Sancho, L.; Matsunaga, N.; Pache, L.; Burgstaller-Muehlbacher, S.; De Jesus, P.D.; Teriete, P.; Hull, M.V.; et al. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 2020, 586, 113–119. [Google Scholar] [CrossRef]
- Bassetti, M.; Vena, A.; Giacobbe, D.R. The novel Chinese coronavirus (2019-nCoV) infections: Challenges for fighting the storm. Eur. J. Clin. Investig. 2020, 50, e13209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehury, B.; Raina, V.; Misra, N.; Suar, M. Effect of mutation on structure, function and dynamics of receptor binding domain of human SARS-CoV-2 with host cell receptor ACE2: A molecular dynamics simulations study. J. Biomol. Struct. Dyn. 2020, 39, 1–15. [Google Scholar] [CrossRef]
- He, Y.; Zhou, Y.; Liu, S.; Kou, Z.; Li, W.; Farzan, M.; Jiang, S. Receptor-binding domain of SARS-CoV spike protein induces highly potent neutralizing antibodies: Implication for developing subunit vaccine. Biochem. Biophys. Res. Commun. 2004, 324, 773–781. [Google Scholar] [CrossRef]
- Sternberg, A.; Naujokat, C. Structural features of coronavirus SARS-CoV-2 spike protein: Targets for vaccination. Life Sci. 2020, 118056. [Google Scholar] [CrossRef] [PubMed]
- Follis, K.E.; York, J.; Nunberg, J.H. Furin cleavage of the SARS coronavirus spike glycoprotein enhances cell–cell fusion but does not affect virion entry. Virology 2006, 350, 358–369. [Google Scholar] [CrossRef] [Green Version]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, M.I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Brink, E.N.; Ter Meulen, J.; Cox, F.; Jongeneelen, M.A.; Thijsse, A.; Throsby, M.; Marissen, W.E.; Rood, P.M.; Bakker, A.B.; Gelderblom, H.R.; et al. Molecular and biological characterization of human monoclonal antibodies binding to the spike and nucleocapsid proteins of severe acute respiratory syndrome coronavirus. J. Virol. 2005, 79, 1635–1644. [Google Scholar] [CrossRef] [Green Version]
- ter Meulen, J.; van den Brink, E.N.; Poon, L.L.; Marissen, W.E.; Leung, C.S.; Cox, F.; Cheung, C.Y.; Bakker, A.Q.; Bogaards, J.A.; van Deventer, E.; et al. Human monoclonal antibody combination against SARS coronavirus: Synergy and coverage of escape mutants. PLoS Med. 2006, 3, e237. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.; Lan, P.D.; Nissley, D.A.; O’Brien, E.A.-O.; Li, M.A.-O. Electrostatic Interactions Explain the Higher Binding Affinity of the CR3022 Antibody for SARS-CoV-2 than the 4A8 Antibody. J. Phys. Chem. B 2021, 125, 7368–7379. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Wu, N.C.; Zhu, X.; Lee, C.-C.D.; So, R.T.; Lv, H.; Mok, C.K.; Wilson, I.A. A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science 2020, 368, 630–633. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Wu, X.; Ren, J.; Zhang, X.; Wang, Y.; Li, C.; Xu, W.; Li, J.; Li, G.; Zheng, W.; et al. Mechanistic Insights to the Binding of Antibody CR3022 Against RBD from SARS-CoV and HCoV-19/SARS-CoV-2: A Computational Study. Comb. Chem. High Throughput Screen. 2021, 24, 1069–1082. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, T.A.-O.; Zhang, J.; Shao, B.; Gong, H.; Wang, Y.; He, X.; Liu, S.; Liu, T.Y. Exploring the Regulatory Function of the N-terminal Domain of SARS-CoV-2 Spike Protein through Molecular Dynamics Simulation. Biomedicines 2021, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Planas, D.; Bruel, T.; Grzelak, L.; Guivel-Benhassine, F.; Staropoli, I.; Porrot, F.; Planchais, C.; Buchrieser, J.; Rajah, M.M.; Bishop, E. Sensitivity of infectious SARS-CoV-2 B. 1.1. 7 and B. 1.351 variants to neutralizing antibodies. Nat. Med. 2021, 27, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Nirula, A.; Heller, B.; Gottlieb, R.L.; Boscia, J.; Morris, J.; Huhn, G.; Cardona, J.; Mocherla, B.; Stosor, V.; et al. SARS-CoV-2 Neutralizing Antibody LY-CoV555 in Outpatients with Covid-19. N. Engl. J. Med. 2021, 384, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.E.; Brown-Augsburger, P.L.; Corbett, K.S.; Westendorf, K.; Davies, J.; Cujec, T.P.; Wiethoff, C.M.; Blackbourne, J.L.; Heinz, B.A.; Foster, D.; et al. The neutralizing antibody, LY-CoV555, protects against SARS-CoV-2 infection in nonhuman primates. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef]
- Yang, Z.; Yang, G.; Zhou, L. Mutation effects of neuraminidases and their docking with ligands: A molecular dynamics and free energy calculation study. J. Comput. Aided Mol. Des. 2013, 27, 935–950. [Google Scholar] [CrossRef]
- Spassov, V.Z.; Yan, L.; Szalma, S. Introducing an Implicit Membrane in Generalized Born/Solvent Accessibility Continuum Solvent Models. J. Phys. Chem. B 2002, 106, 8726–8738. [Google Scholar] [CrossRef]
- Spassov, V.Z.; Yan, L. A fast and accurate computational approach to protein ionization. Protein. Sci. 2008, 17, 1955–1970. [Google Scholar] [CrossRef] [PubMed]
- Spassov, V.Z.; Yan, L. pH-selective mutagenesis of protein–protein interfaces: In silico design of therapeutic antibodies with prolonged half-life. Proteins Struct. Funct. Bioinform. 2013, 81, 704–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prigent, J.; Jarossay, A.; Planchais, C.; Eden, C.; Dufloo, J.; Kok, A.; Lorin, V.; Vratskikh, O.; Couderc, T.; Bruel, T.; et al. Conformational Plasticity in Broadly Neutralizing HIV-1 Antibodies Triggers Polyreactivity. Cell Rep. 2018, 23, 2568–2581. [Google Scholar] [CrossRef] [Green Version]
- Accelrys Discovery Studio 4.5, Version 4.5. 2015. Available online: http://accelrys.com (accessed on 6 November 2021).
- Jiayi, R.; Zhiwei, Y.; Nianjue, Z.; Junqi, L.; Chunlong, Y.; Shujian, L.; Bing, Y.; Junqing, H.; Huaxin, L.; Xiaohui, Y. Effect of force fields and water models on EGFRvIII-MR1 (scFv) complex by molecular dynamics simulation, MM-PBSA calculation, and ITC experiment. Chem. J. Chin. Univ. 2017, 11, 2070–2076. [Google Scholar]
- Yuzlenko, O.; Lazaridis, T. Membrane protein native state discrimination by implicit membrane models. J. Comput. Chem. 2013, 34, 731–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Chen, C.-F.; Zhao, B.-B.; Gong, L.-L.; Jin, W.-J.; Liu, J.-J.; Wang, J.-F.; Wang, T.-T.; Yuan, X.-H.; He, Y.-W. A novel antibody humanization method based on epitopes scanning and molecular dynamics simulation. PLoS ONE 2013, 8, e80636. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.-H.; Qu, Z.-Y.; Wu, X.-M.; Wang, Y.-C.; Wei, F.-X.; Zhang, H.; Liu, L.; Yang, Z. Homology Modeling and Evolution Trace Analysis of Human Adenovirus Type 3 Hexon. Chem. J. Chin. Univ. 2009, 30, 1636. [Google Scholar]
- Chandrasekhar, I.; Kastenholz, M.; Lins, R.D.; Oostenbrink, C.; Schuler, L.D.; Tieleman, D.P.; van Gunsteren, W.F. A consistent potential energy parameter set for lipids: Dipalmitoylphosphatidylcholine as a benchmark of the GROMOS96 45A3 force field. Eur. Biophys. J. 2003, 32, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.L.; Pei, G.K. Modification of a PCRBased Site-Directed Mutagenesis Method. Biotechniques 1997, 23, 570–574. [Google Scholar] [CrossRef]
- Gapsys, V.; Michielssens, S.; Seeliger, D.; de Groot, B.L. pmx: Automated protein structure and topology generation for alchemical perturbations. J. Comput. Chem. 2015, 15, 348–354. [Google Scholar] [CrossRef] [Green Version]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Interaction Models for Water in Relation to Protein Hydration. In Intermolecular Forces; Pullman, B., Ed.; Springer: Dordrecht, The Netherland, 1981; Volume 14. [Google Scholar]
- Zhang, Y.N.; Zhang, X.C.; Zhu, R.; Yao, W.C.; Xu, J.W.; Wang, M.; Ren, J.Y.; Xu, C.Z.; Huang, Z.R.; Zhang, X.W.; et al. Computational and Experimental Approaches to Decipher the Binding Mechanism of General Odorant-Binding Protein 2 from Athetis lepigone to Chlorpyrifos and Phoxim. J. Agric. Food Chem. 2021, 69, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Delhommelle, J.; Evans, D.J. Comparison of thermostatting mechanisms in NVT and NPT simulations of decane under shear. J. Chem. Phys. 2001, 115, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Makov, G.; Payne, M. Periodic boundary conditions in ab initio calculations. Phys. Rev. B 1995, 51, 4014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Yuan, X.; Wang, Y.; Qu, Z.; Ren, J.; Wu, X.; Wang, J. Phylogenetic and structural analysis of major surface proteins hemagglutinin and neuraminidase of novel avian influenza virus A H7N9 from chinese patient. Chem. Res. Chin. Univ. 2013, 29, 934–940. [Google Scholar] [CrossRef]
- Grubmüller, H.; Heller, H.; Windemuth, A.; Schulten, K. Generalized Verlet algorithm for efficient molecular dynamics simulations with long-range interactions. Mol. Simul. 1991, 6, 121–142. [Google Scholar] [CrossRef]
- Ren, J.; Huang, J.; Yang, B.; Lin, S.; Li, J.; Liao, H.; Yuan, X.; Wu, X.; Ou, S. Docking and molecular dynamics: Simulation of the inhibition of H5N1 influenza virus (Anhui 2005) neuraminidase (NA) by chlorogenic acid (CHA). Int. J. Clin. Exp. Med. 2019, 12, 9815–9823. [Google Scholar]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Modeling 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.-H.; Wang, Y.-C.; Qu, Z.; Ren, J.; Wang, J.; Guo, Y.-Y.; Wang, Y.-X.; Hua, D.; Wu, X.-M.; Yang, Z.-W. Novel Rapid Molecular Modeling Method Based on Evolutional Tree for Human Adenovirus Hexon Proteins Family. Chem. J. Chin. Univ. 2011, 32, 1838. [Google Scholar]
- Gohlke, H.; Case, D.A. Converging free energy estimates: MM-PB(GB)SA studies on the protein-protein complex Ras-Raf. J Comput. Chem. 2004, 25, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.K.; Koča, J. Assessing the performance of MM/PBSA, MM/GBSA, and QM–MM/GBSA approaches on protein/carbohydrate complexes: Effect of implicit solvent models, QM methods, and entropic contributions. J. Phys. Chem. B 2018, 122, 8113–8121. [Google Scholar] [CrossRef]
- Horvath, D. A virtual screening approach applied to the search for trypanothione reductase inhibitors. J. Med. Chem. 1997, 40, 2412–2423. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Liu, X.; Zhang, J.Z. Interaction entropy: A new paradigm for highly efficient and reliable computation of protein–ligand binding free energy. J. Am. Chem. Soc. 2016, 138, 5722–5728. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Mutation Sites (IMGT Number Scheme) | ΔGbind (kJ/mol) | |
|---|---|---|---|
| 6W41 | N76200 | ||
| Heavy Chain CDR1 | G29W | −1.54 | −1.29 |
| T36F | −1.11 | −2.48 | |
| T36Y | −1.07 | −1.14 | |
| Heavy Chain CDR3 | S103R | −2.21 | −2.96 |
| S103I | −1.84 | −1.25 | |
| S103F | −2.05 | −2.04 | |
| S103W | −1.35 | −1.87 | |
| S103Y | −3.13 | −2.43 | |
| Light Chain CDR1 | S33R | −2.62 | −2.94 |
| S33H | −1.34 | −2.41 | |
| S33I | −1.69 | −2.13 | |
| S33L | −2.31 | −2.75 | |
| S33K | −1.96 | −1.67 | |
| S33M | −1.47 | −1.47 | |
| S33F | −1.81 | −2.92 | |
| S33W | −2.38 | −2.95 | |
| S33Y | −2.35 | −2.29 | |
| N35Y | −2.01 | −1.48 | |
| Analyte Solution | Capture Solution | Ka (M−1s−1) | Kd (s−1) | KD (M) |
|---|---|---|---|---|
| SARS-CoV RBD | CR3022 | 2.44 × 105 | 5.27 × 10−4 | 2.16 × 10−9 |
| S103F(H)–S33R(L) | 6.45 × 105 | 4.58 × 10−4 | 7.10 × 10−10 | |
| S103Y(H)–S33R(L) | 6.91 × 105 | 5.35 × 10−4 | 7.75 × 10−10 | |
| SARS-CoV–2 RBD | CR3022 | 1.17 × 106 | 1.74 × 10−2 | 1.49 × 10−8 |
| S103F(H)–S33R(L) | 2.28 × 106 | 2.28 × 10−3 | 9.99 × 10−10 | |
| S103Y(H)–S33R(L) | 2.15 × 106 | 2.47 × 10−3 | 1.15 × 10−9 |
| Complex | Van der Waal Energy (ΔGvdw) | Electrostatic Energy (ΔGele) | Polar Solvation Energy (ΔGPB) | SASA Energy (ΔGSA) | Binding Energy (ΔGbind) |
|---|---|---|---|---|---|
| SARS-CoV-2 RBD–CR3022 | −401.25 ± 2.59 | −261.41 ± 3.35 | 778.63 ± 12.93 | −46.18 ± 0.38 | −290.01 ± 20.57 |
| SARS-CoV-2 RBD–S103F–S33R | −451.37 ± 2.62c | −328.87 ± 4.42 | 905.99 ± 13.23 | −50.98 ± 0.37 | −321.63 ± 23.97 |
| SARS-CoV-2 RBD–S103Y–S33R | −445.01 ± 2.88 | −303.79 ± 2.89 | 825.26 ± 8.59 | −51.87 ± 0.38 | −348.21 ± 17.85 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, W.; Zhong, N.; Li, X.; Ren, J.; Wang, Y.; Li, C.; Yao, G.; Zhu, R.; Wang, X.; Jia, Z.; et al. Structure Based Affinity Maturation and Characterizing of SARS-CoV Antibody CR3022 against SARS-CoV-2 by Computational and Experimental Approaches. Viruses 2022, 14, 186. https://doi.org/10.3390/v14020186
Yu W, Zhong N, Li X, Ren J, Wang Y, Li C, Yao G, Zhu R, Wang X, Jia Z, et al. Structure Based Affinity Maturation and Characterizing of SARS-CoV Antibody CR3022 against SARS-CoV-2 by Computational and Experimental Approaches. Viruses. 2022; 14(2):186. https://doi.org/10.3390/v14020186
Chicago/Turabian StyleYu, Wei, Nan Zhong, Xin Li, Jiayi Ren, Yueming Wang, Chengming Li, Gui Yao, Rui Zhu, Xiaoli Wang, Zhenxing Jia, and et al. 2022. "Structure Based Affinity Maturation and Characterizing of SARS-CoV Antibody CR3022 against SARS-CoV-2 by Computational and Experimental Approaches" Viruses 14, no. 2: 186. https://doi.org/10.3390/v14020186
APA StyleYu, W., Zhong, N., Li, X., Ren, J., Wang, Y., Li, C., Yao, G., Zhu, R., Wang, X., Jia, Z., Wu, C., Chen, R., Zheng, W., Liao, H., Wu, X., & Yuan, X. (2022). Structure Based Affinity Maturation and Characterizing of SARS-CoV Antibody CR3022 against SARS-CoV-2 by Computational and Experimental Approaches. Viruses, 14(2), 186. https://doi.org/10.3390/v14020186