
Inspecting the Ribozyme Region of Hepatitis Delta Virus Genotype 1: Conservation and Variability

 , ,
, ,  , , , , , , ,
, , , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Samples
2.2. Serologic and Molecular Assays
2.3. Next-Generation Sequencing of the Ribozyme Region
2.4. Quasispecies Analysis and Statistics
2.5. In Vitro Test of Mutations
3. Results
3.1. Patients and Sequencing
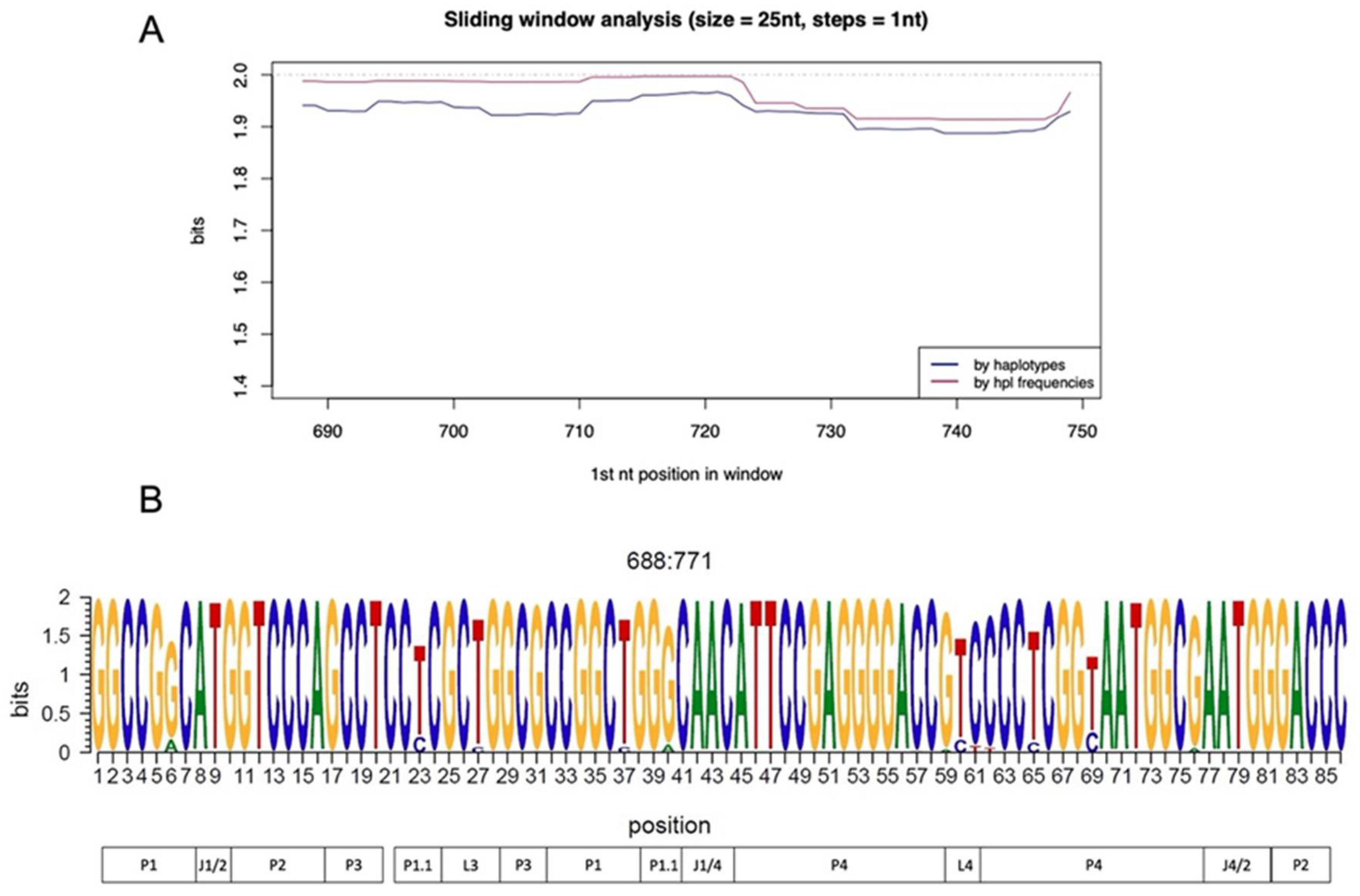
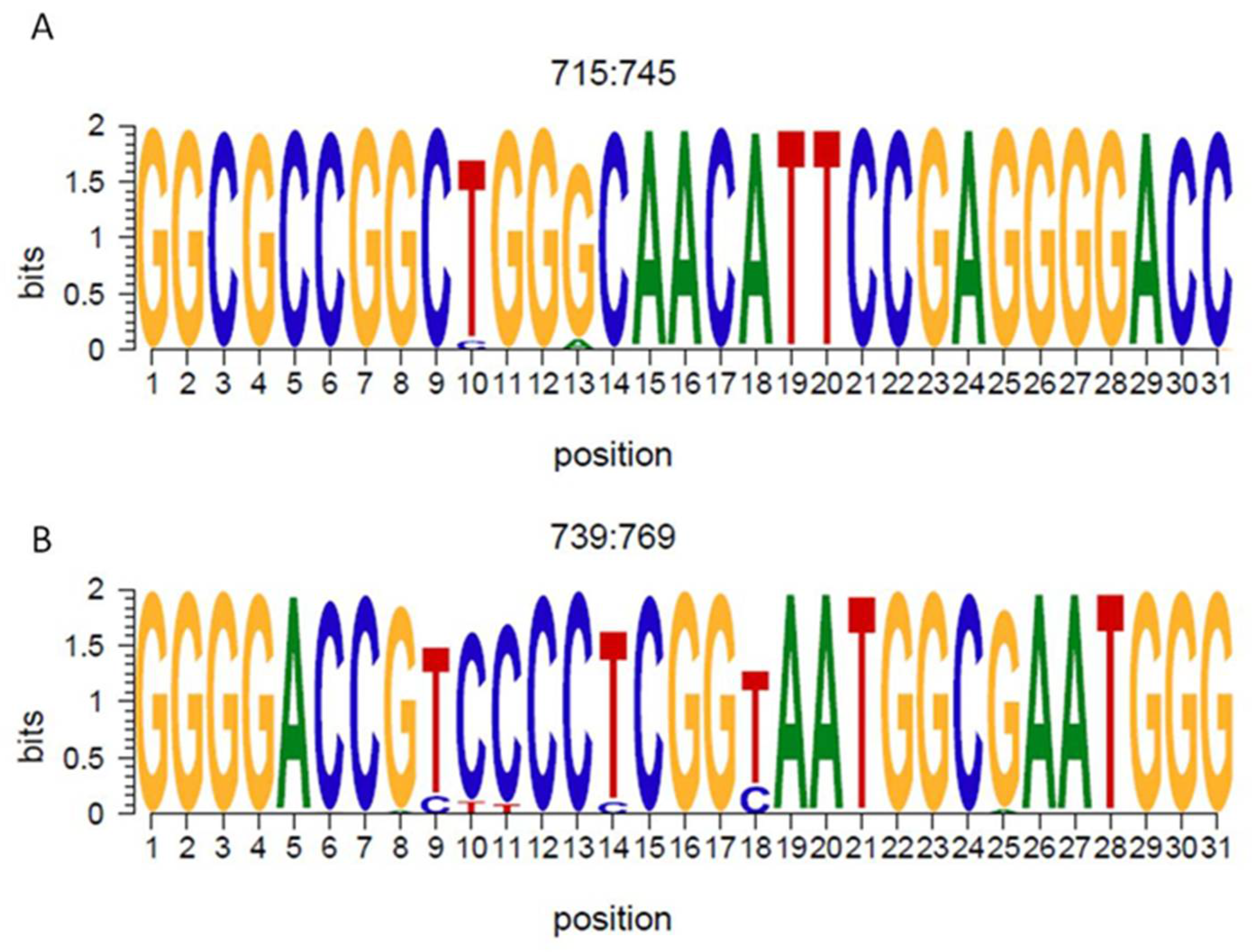
3.2. Ribozyme Conservation
3.3. Ribozyme Quasispecies (QS) Evolution and Variability during Follow-Up
3.4. Analysis of Mutations
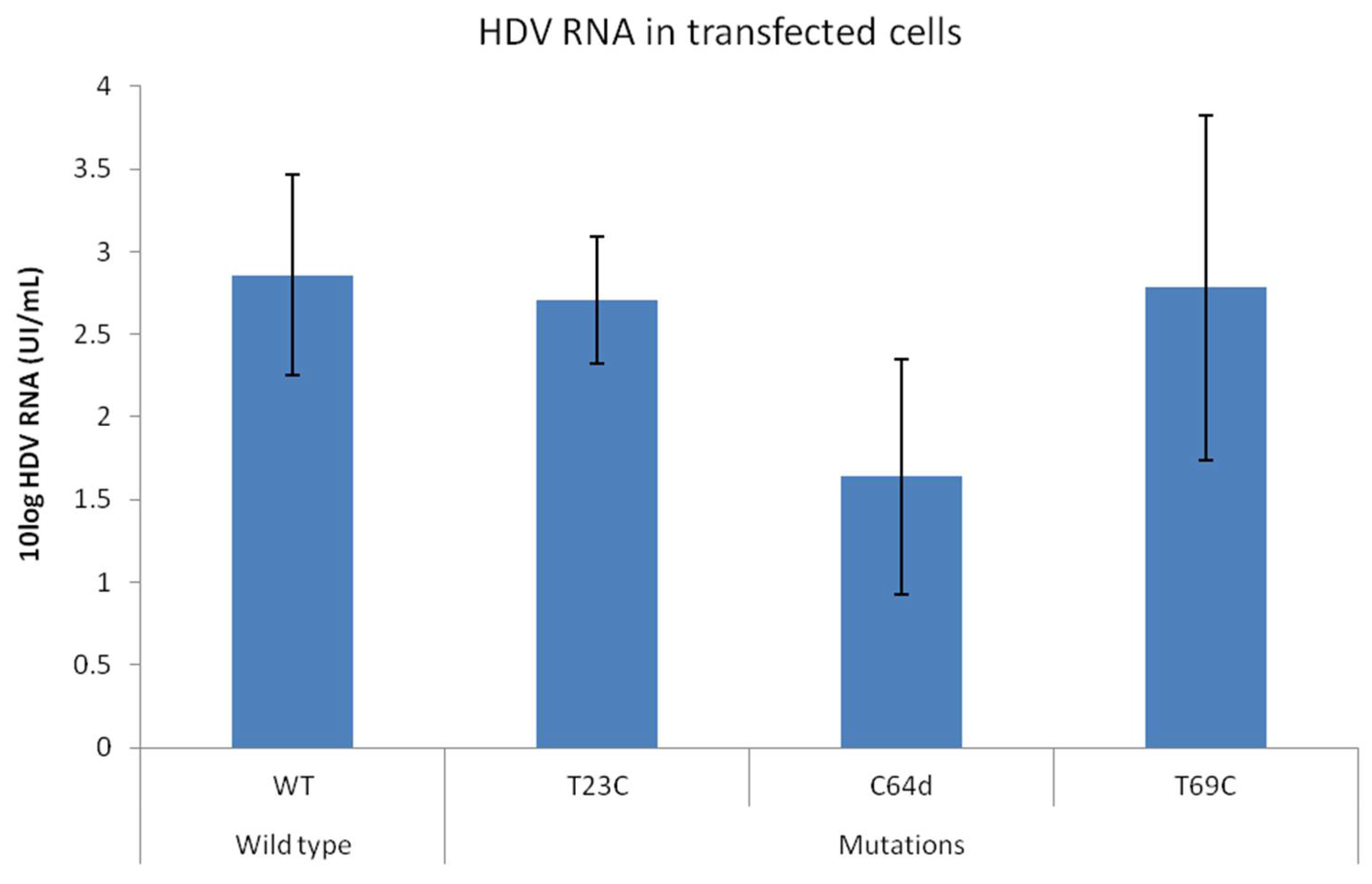
3.5. In Vitro Test of Mutations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sureau, C.; Negro, F. The Hepatitis Delta Virus: Replication and Pathogenesis. J. Hepatol. 2016, 64, S102–S116. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.-Y.; Shen, D.-T.; Ji, D.-Z.; Han, P.-C.; Zhang, W.-M.; Ma, J.-F.; Chen, W.-S.; Goyal, H.; Pan, S.; Xu, H.-G. Prevalence and Burden of Hepatitis D Virus Infection in the Global Population: A Systematic Review and Meta-Analysis. Gut 2019, 68, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Wedemeyer, H.; Negro, F. Devil Hepatitis D: An Orphan Disease or Largely Underdiagnosed? Gut 2019, 68, 381–382. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.-C.; Chao, M. Molecular Cloning and Expression of the Hepatitis Delta Virus Genotype IIb Genome. Biochem. Biophys. Res. Commun. 2003, 303, 357–363. [Google Scholar] [CrossRef]
- Farci, P.; Anna Niro, G. Current and Future Management of Chronic Hepatitis D. Gastroenterol. Hepatol. 2018, 14, 342–351. [Google Scholar]
- Sharmeen, L.; Kuo, M.Y.; Dinter-Gottlieb, G.; Taylor, J. Antigenomic RNA of Human Hepatitis Delta Virus Can Undergo Self-Cleavage. J. Virol. 1988, 62, 2674–2679. [Google Scholar] [CrossRef] [Green Version]
- Reid, C.E.; Lazinski, D.W. A Host-Specific Function Is Required for Ligation of a Wide Variety of Ribozyme-Processed RNAs. Proc. Natl. Acad. Sci. USA 2000, 97, 424–429. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.N.; Lin, Y.J.; Lin, F.P.; Makino, S.; Chang, M.F.; Lai, M.M. Human Hepatitis Delta Virus RNA Subfragments Contain an Autocleavage Activity. Proc. Natl. Acad. Sci. USA 1989, 86, 1831–1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharmeen, L.; Kuo, M.Y.; Taylor, J. Self-Ligating RNA Sequences on the Antigenome of Human Hepatitis Delta Virus. J. Virol. 1989, 63, 1428–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruger, K.; Grabowski, P.J.; Zaug, A.J.; Sands, J.; Gottschling, D.E.; Cech, T.R. Self-Splicing RNA: Autoexcision and Autocyclization of the Ribosomal RNA Intervening Sequence of Tetrahymena. Cell 1982, 31, 147–157. [Google Scholar] [CrossRef]
- Golden, B.L. Two Distinct Catalytic Strategies in the Hepatitis Delta Virus Ribozyme Cleavage Reaction. Biochemistry 2011, 50, 9424–9433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Breaker, R.R. Structural Diversity of Self-Cleaving Ribozymes. Proc. Natl. Acad. Sci. USA 2000, 97, 5784–5789. [Google Scholar] [CrossRef] [Green Version]
- Ferré-D’Amaré, A.R.; Zhou, K.; Doudna, J.A. Crystal Structure of a Hepatitis Delta Virus Ribozyme. Nature 1998, 395, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Riccitelli, N.; Lupták, A. HDV Family of Self-Cleaving Ribozymes. Prog. Mol. Biol. Transl. Sci. 2013, 120, 123–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thill, G.; Vasseur, M.; Tanner, N.K. Structural and Sequence Elements Required for the Self-Cleaving Activity of the Hepatitis Delta Virus Ribozyme. Biochemistry 1993, 32, 4254–4262. [Google Scholar] [CrossRef] [PubMed]
- Been, M.D.; Wickham, G.S. Self-Cleaving Ribozymes of Hepatitis Delta Virus RNA. Eur. J. Biochem. 1997, 247, 741–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quer, J.; Rodríguez-Frias, F.; Gregori, J.; Tabernero, D.; Soria, M.E.; García-Cehic, D.; Homs, M.; Bosch, A.; Pintó, R.M.; Esteban, J.I.; et al. Deep Sequencing in the Management of Hepatitis Virus Infections. Virus Res. 2017, 239, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Perales, C. Viral Quasispecies. PLoS Genet. 2019, 15, e1008271. [Google Scholar] [CrossRef] [Green Version]
- Orito, E.; Mizokami, M.; Ina, Y.; Moriyama, E.N.; Kameshima, N.; Yamamoto, M.; Gojobori, T. Host-Independent Evolution and a Genetic Classification of the Hepadnavirus Family Based on Nucleotide Sequences. Proc. Natl. Acad. Sci. USA 1989, 86, 7059–7062. [Google Scholar] [CrossRef] [Green Version]
- Revill, P.A.; Tu, T.; Netter, H.J.; Yuen, L.K.W.; Locarnini, S.A.; Littlejohn, M. The Evolution and Clinical Impact of Hepatitis B Virus Genome Diversity. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 618–634. [Google Scholar] [CrossRef]
- Tabernero, D.; Cortese, M.F.; Buti, M.; Rodriguez-Frias, F. HDV Evolution—Will Viral Resistance Be an Issue in HDV Infection? Curr. Opin. Virol. 2018, 32, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Homs, M.; Rodriguez-Frias, F.; Gregori, J.; Ruiz, A.; Reimundo, P.; Casillas, R.; Tabernero, D.; Godoy, C.; Barakat, S.; Quer, J.; et al. Evidence of an Exponential Decay Pattern of the Hepatitis Delta Virus Evolution Rate and Fluctuations in Quasispecies Complexity in Long-Term Studies of Chronic Delta Infection. PLoS ONE 2016, 11, e0158557. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-M.; Bih, F.-Y.; Chao, Y.-C.; Govindarajan, S.; Lai, M.M.C. Evolution of Hepatitis Delta Virus Rna during Chronic Infection. Virology 1992, 188, 265–273. [Google Scholar] [CrossRef]
- le Gal, F.; Gault, E.; Ripault, M.-P.; Serpaggi, J.; Trinchet, J.-C.; Gordien, E.; Deny, P. Eighth Major Clade for Hepatitis Delta Virus. Emerg. Infect. Dis. 2006, 12, 1447–1450. [Google Scholar] [CrossRef] [PubMed]
- Dény, P. Hepatitis Delta Virus Genetic Variability: From Genotypes I, II, III to Eight Major Clades? In Hepatitis Delta Virus; Springer: Berlin/Heidelberg, Germany, 2006; pp. 151–171. [Google Scholar]
- Homs, M.; Caballero, A.; Gregori, J.; Tabernero, D.; Quer, J.; Nieto, L.; Esteban, R.; Buti, M.; Rodriguez-Frias, F. Clinical Application of Estimating Hepatitis B Virus Quasispecies Complexity by Massive Sequencing: Correlation between Natural Evolution and On-Treatment Evolution. PLoS ONE 2014, 9, e112306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sopena, S.; Godoy, C.; Tabernero, D.; Homs, M.; Gregori, J.; Riveiro-Barciela, M.; Ruiz, A.; Esteban, R.; Buti, M.; Rodríguez-Frías, F. Quantitative Characterization of Hepatitis Delta Virus Genome Edition by Next-Generation Sequencing. Virus Res. 2018, 243, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garcia, S.; Cortese, M.F.; Rodríguez-Algarra, F.; Tabernero, D.; Rando-Segura, A.; Quer, J.; Buti, M.; Rodríguez-Frías, F. Next-Generation Sequencing for the Diagnosis of Hepatitis B: Current Status and Future Prospects. Expert Rev. Mol. Diagn. 2021, 21, 381–396. [Google Scholar] [CrossRef]
- Lempp, F.A.; Ni, Y.; Urban, S. Hepatitis Delta Virus: Insights into a Peculiar Pathogen and Novel Treatment Options. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Scheller, L.; Hilgard, G.; Anastasiou, O.; Dittmer, U.; Kahraman, A.; Wedemeyer, H.; Deterding, K. Poor Clinical and Virological Outcome of Nucleos(t)Ide Analogue Monotherapy in HBV/HDV Co-Infected Patients. Medicine 2021, 100, e26571. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Syed, Y.Y. Bulevirtide: First Approval. Drugs 2020, 80, 1601–1605. [Google Scholar] [CrossRef] [PubMed]
- Abbas, Z. Management of Hepatitis Delta: Need for Novel Therapeutic Options. World J. Gastroenterol. 2015, 21, 9461. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-Y.; Wu, J.-C.; Chiang, T.-Y.; Huang, Y.-H.; Su, C.-W.; Sheen, I.-J. Positive Selection of Hepatitis Delta Antigen in Chronic Hepatitis D Patients. J. Virol. 2007, 81, 4438–4444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul-Ehrlich-Institut A WHO Collaborating Centre Bundesinstitut Für Impfstoffe Und Biomedizinische Arzneimittel for Quality Assurance of Blood Products and Federal Institute for Vaccines and Biomedicines in Vitro Diagnostic Devices. Available online: https://www.pei.de/SharedDocs/Downloads/EN/regulation-en/referencematerial/7657-12-ifu.pdf?__blob=publicationFile&v=2 (accessed on 22 January 2016).
- R Core Team (2020)—European Environment Agency. Available online: https://www.eea.europa.eu/data-and-maps/indicators/oxygen-consuming-substances-in-rivers/r-development-core-team-2006 (accessed on 9 January 2022).
- Cortese, M.F.; González, C.; Gregori, J.; Casillas, R.; Carioti, L.; Guerrero-Murillo, M.; Riveiro-Barciela, M.; Godoy, C.; Sopena, S.; Yll, M.; et al. Sophisticated Viral Quasispecies with a Genotype-Related Pattern of Mutations in the Hepatitis B X Gene of HBeAg-ve Chronically Infected Patients. Sci. Rep. 2021, 11, 4215. [Google Scholar] [CrossRef]
- Godoy, C.; Tabernero, D.; Sopena, S.; Gregori, J.; Francesca Cortese, M.; González, C.; Casillas, R.; Yll, M.; Rando, A.; López-Martínez, R.; et al. Characterization of Hepatitis B Virus X Gene Quasispecies Complexity in Mono-Infection and Hepatitis Delta Virus Superinfection. World J. Gastroenterol. 2019, 25, 1566–1579. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.T.; Netter, H.J.; Lazinski, D.W.; Taylor, J.M. Effects of Nucleotide Changes on the Ability of Hepatitis Delta Virus to Transcribe, Process, and Accumulate Unit-Length, Circular RNA. J. Virol. 1997, 71, 5408–5414. [Google Scholar] [CrossRef] [Green Version]
- Homs, M.; Buti, M.; Quer, J.; Jardi, R.; Schaper, M.; Tabernero, D.; Ortega, I.; Sanchez, A.; Esteban, R.; Rodriguez-Frias, F. Ultra-Deep Pyrosequencing Analysis of the Hepatitis B Virus PreCore Region and Main Catalytic Motif of the Viral Polymerase in the Same Viral Genome. Nucleic Acids Res. 2011, 39, 8457–8471. [Google Scholar] [CrossRef] [PubMed]
- Yll, M.; Cortese, M.F.; Guerrero-Murillo, M.; Orriols, G.; Gregori, J.; Casillas, R.; González, C.; Sopena, S.; Godoy, C.; Vila, M.; et al. Conservation and Variability of Hepatitis B Core at Different Chronic Hepatitis Stages. World J. Gastroenterol. 2020, 26, 2584–2598. [Google Scholar] [CrossRef]
- Chang, M.F.; Chen, C.H.; Lin, S.L.; Chen, C.J.; Chang, S.C. Functional Domains of Delta Antigens and Viral RNA Required for RNA Packaging of Hepatitis Delta Virus. J. Virol. 1995, 69, 2508–2514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirvani-Dastgerdi, E.; Amini-Bavil-Olyaee, S.; Alavian, S.M.; Trautwein, C.; Tacke, F. Comprehensive Analysis of Mutations in the Hepatitis Delta Virus Genome Based on Full-Length Sequencing in a Nationwide Cohort Study and Evolutionary Pattern during Disease Progression. Clin. Microbiol. Infect. 2015, 21, 510.e11–510.e23. [Google Scholar] [CrossRef] [Green Version]
- Mentha, N.; Clément, S.; Negro, F.; Alfaiate, D. A Review on Hepatitis D: From Virology to New Therapies. J. Adv. Res. 2019, 17, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, D.; Castelnau, C.; Tout, I.; Boyer, N.; Narguet, S.; Menasria Benazzouz, S.; Louis, Z.; Pons-Kerjean, N.; Giuly, N.; Marcellin, P.; et al. New Therapies for Hepatitis Delta Virus Infection. Liver Int. 2021, 41, 30–37. [Google Scholar] [CrossRef]
- Asselah, T.; Loureiro, D.; le Gal, F.; Narguet, S.; Brichler, S.; Bouton, V.; Abazid, M.; Boyer, N.; Giuly, N.; Gerber, A.; et al. Early Virological Response in Six Patients with Hepatitis D Virus Infection and Compensated Cirrhosis Treated with Bulevirtide in Real-Life. Liver Int. 2021, 41, 1509–1517. [Google Scholar] [CrossRef]
- Chang, J.; Taylor, J.M. Susceptibility of Human Hepatitis Delta Virus RNAs to Small Interfering RNA Action. J. Virol. 2003, 77, 9728–9731. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Mao, Q.; Li, Q. Inhibitory Effect of Replication and Expression of HDV by Antisense Oligodeoxynucleotides in H1 Delta 9 Cell. Chin. J. Hepatol. 1999, 7, 13–14. [Google Scholar]
- Ye, X.; Tateno, C.; Thi, E.P.; Kakuni, M.; Snead, N.M.; Ishida, Y.; Barnard, T.R.; Sofia, M.J.; Shimada, T.; Lee, A.C.H. Hepatitis B Virus Therapeutic Agent ARB-1740 Has Inhibitory Effect on Hepatitis Delta Virus in a New Dually-Infected Humanized Mouse Model. ACS Infect. Dis. 2019, 5, 738–749. [Google Scholar] [CrossRef] [PubMed]
- Chad, Y.-C.; Chang, M.-F.; Gust, I.; Lai, M.M.C. Sequence Conservation and Divergence of Hepatitis δ Virus RNA. Virology 1990, 178, 384–392. [Google Scholar] [CrossRef]
- Rosenstein, S.P.; Been, M.D. Hepatitis Delta Virus Ribozymes Fold to Generate a Solvent-Inaccessible Core with Essential Nucleotides Near the Cleavage Site Phosphate. Biochemistry 1996, 35, 11403–11413. [Google Scholar] [CrossRef] [PubMed]
- Perrotta, A.T.; Been, M.D. A Pseudoknot-like Structure Required for Efficient Self-Cleavage of Hepatitis Delta Virus RNA. Nature 1991, 350, 434–436. [Google Scholar] [CrossRef] [PubMed]
- Chadalavada, D.M.; Cerrone-Szakal, A.L.; Bevilacqua, P.C. Wild-Type Is the Optimal Sequence of the HDV Ribozyme under Cotranscriptional Conditions. RNA 2007, 13, 2189–2201. [Google Scholar] [CrossRef] [Green Version]
- Perrotta, A.T.; Been, M.D. Core Sequences and a Cleavage Site Wobble Pair Required for HDV Antigenomic Ribozyme Self-Cleavage. Nucleic Acids Res. 1996, 24, 1314–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanner, N.K.; Schaff, S.; Thill, G.; Petit-Koskas, E.; Crain-Denoyelle, A.-M.; Westhof, E. A Three-Dimensional Model of Hepatitis Delta Virus Ribozyme Based on Biochemical and Mutational Analyses. Curr. Biol. 1994, 4, 488–498. [Google Scholar] [CrossRef]
- Timm, J.; Lauer, G.M.; Kavanagh, D.G.; Sheridan, I.; Kim, A.Y.; Lucas, M.; Pillay, T.; Ouchi, K.; Reyor, L.L.; zur Wiesch, J.S.; et al. CD8 Epitope Escape and Reversion in Acute HCV Infection. J. Exp. Med. 2004, 200, 1593–1604. [Google Scholar] [CrossRef] [PubMed]
- Leslie, A.J.; Pfafferott, K.J.; Chetty, P.; Draenert, R.; Addo, M.M.; Feeney, M.; Tang, Y.; Holmes, E.C.; Allen, T.; Prado, J.G.; et al. HIV Evolution: CTL Escape Mutation and Reversion after Transmission. Nat. Med. 2004, 10, 282–289. [Google Scholar] [CrossRef]
- Cotrina, M.; Buti, M.; Jardi, R.; Quer, J.; Rodriguez, F.; Pascual, C.; Esteban, R.; Guardia, J. Hepatitis Delta Genotypes in Chronic Delta Infection in the Northeast of Spain (Catalonia). J. Hepatol. 1998, 28, 971–977. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amplification Step | Primer | Amplified Region | Primer Sequence (5′→3′) | Protocol |
|---|---|---|---|---|
| RT | RT rv | 1435–1454 | TGGCTGGGAAACATCAAAGG | RT 42 °C 60 min; inactivation 70 °C 10 min; cooling 20 °C ∞ |
| 1st PCR | 1a fw | 1435–1454 | TGGCTGGGAAACATCAAAGG | 95 °C 1 min; (94 °C 20 s, 54 °C 20 s, 72 °C 45 s) × 40 cycles; 72 °C 3 min |
| 1a rv | 308–326 | CCTCCAGAGGACCCCTTCA | ||
| M13 PCR | M13-fw | 883–900 | CACAGGAAACAGCTATGACCTCGGCATGGCATCTCCAC | 95° C 2 min; (94 °C 20 s, 60 °C 20 s, 72 °C 30 s) × 35 cycles; 72 °C 3 min |
| M13-rv | 663–683 | GTTGTAAAACGACGGCCAGTCGCGTTCCATCCTTTCTTACC | ||
| MID PCR | MID fw | - | MID-GTTGTAAAACGACGGCCAGT | 95 °C 2 min; (94 °C 20 s, 60 °C 20 s, 72 °C 45 s) × 25 cycles; 72 °C 3 min |
| MID rv | - | MID-CACAGGAAACAGCTATGACC |
| Markers | Sample A | Sample B | p |
|---|---|---|---|
| HDV RNA Median (IQR) Log10 (IU/mL) | 5.76 (5.02–5.83) | 5.76 (3.68–5.76) | 0.383 |
| AST (UI/L) Median (IQR) | 88 (45–133) | 90 (36.25–126) | 0.551 |
| ALT (UI/L) Median (IQR) | 100 (58–158.5) | 89.25 (44.5–133.75) | 0.439 |
| PLATELETS (UI/L) Median (IQR) | 89 (123–212) | 79 (118–197) | 0.966 |
| HBV-DNA (IU/mL) | Low/IND | Low/IND | |
| HBsAg Median (IQR) Log10 (IU/mL) | 3.96 (3.57–4.11) | 3.92 (3.39–4.03) | 0.827 |
| Mutation | Sample A (%freq) | Sample B (%freq) |
|---|---|---|
| G6A | 0.84 ± 0.43% | 0.83 ± 0.31% |
| T23C | 3.04 ± 2.27% | 5.55 ± 8.91% |
| T27C | 0.12 ± 0.3% | 0.88 ± 3.5% |
| G40A | 0.25 ± 0.22% | 0.41 ± 0.24% |
| G59A | 0.07 ± 0.03% | 0.08 ± 0.02% |
| T60C | 6.15 ± 30.64% | 6.42 ± 35.01% |
| C61T | 0.15 ± 0.24% | 0.14 ± 0.27% |
| C62T | 0.12 ± 0.17% | 0.08 ± 0.12% |
| C64d | 47.17 ± 1.66% | 48.88 ± 2.47% |
| T65C | 6.02 ± 39.66% | 6.29 ± 44.04% |
| T69C | 12.02 ± 21.99% | 12.75 ± 44.04% |
| G76A | 0.53 ± 2.49% | 0.44 ± 17.68% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pacin-Ruiz, B.; Cortese, M.F.; Tabernero, D.; Sopena, S.; Gregori, J.; García-García, S.; Casillas, R.; Najarro, A.; Aldama, U.; Palom, A.; et al. Inspecting the Ribozyme Region of Hepatitis Delta Virus Genotype 1: Conservation and Variability. Viruses 2022, 14, 215. https://doi.org/10.3390/v14020215
Pacin-Ruiz B, Cortese MF, Tabernero D, Sopena S, Gregori J, García-García S, Casillas R, Najarro A, Aldama U, Palom A, et al. Inspecting the Ribozyme Region of Hepatitis Delta Virus Genotype 1: Conservation and Variability. Viruses. 2022; 14(2):215. https://doi.org/10.3390/v14020215
Chicago/Turabian StylePacin-Ruiz, Beatriz, María Francesca Cortese, David Tabernero, Sara Sopena, Josep Gregori, Selene García-García, Rosario Casillas, Adrián Najarro, Unai Aldama, Adriana Palom, and et al. 2022. "Inspecting the Ribozyme Region of Hepatitis Delta Virus Genotype 1: Conservation and Variability" Viruses 14, no. 2: 215. https://doi.org/10.3390/v14020215