Abstract
Fowl adenoviruses (FAdVs) are infectious agents, mainly of chickens, which cause economic losses to the poultry industry. Only a single serotype, namely FAdV-5, constitutes the species Fowl aviadenovirus B (FAdV-B); however, recently, phylogenetic analyses have identified divergent strains of the species, implicating a more complex scenario and possibly a novel serotype. Therefore, field isolates of the species were collected to investigate the contemporary diversification within FAdV-B, including traditional serotyping. Full genomes of fourteen FAdV-B strains were sequenced and four strains, possessing discriminatory mutations in the antigenic domains, were compared using virus cross-neutralization. Essentially, strains with identical antigenic signatures to that of the first described divergent strain were found in the complete new dataset. While chicken antiserum against FAdV-5 reference strain 340 could not neutralize any of the newly isolated viruses, low homologous/heterologous titer ratios were measured reciprocally. Although they argue against a new serotype, our results indicate the emergence of escape variants in FAdV-B. Charge-influencing amino acid substitutions accounted for only a few mutations between the strains; still, these enabled one-way cross-neutralization only. These findings underline the continued merit of the cross-neutralization test as the gold standard for serotyping, complementary to advancing sequence data, and provide a snapshot of the actual diversity and evolution of species FAdV-B.
1. Introduction
Adenoviruses possess an icosahedral, non-enveloped capsid and a linear, non-segmented, double-stranded DNA genome, and infect all classes of vertebrates [1,2]. The family Adenoviridae was recently included in the realm Varidnaviria, kingdom Bamfordvirae, phylum Preplasmiviricota, class Tectiliviricetes and order Rowavirales [3]. Within the family, the members of the genus Aviadenovirus infect exclusively birds [4]. Strains of the species Fowl aviadenovirus A–E can cause severe economic losses for the poultry industry, especially in broiler production [5,6]. These five species are further divided into 12 serotypes (FAdV-1-8a and 8b-11), and there is a single serotype, fowl adenovirus 5 (FAdV-5), classified into the species Fowl aviadenovirus B (FAdV-B) [7,8].
Available clinical data regarding this species and its sole serotype are relatively limited compared to other FAdV species. All other FAdV species contain certain serotypes which are the etiological agents of well-characterized chicken diseases (inclusion body hepatitis, hepatitis–hydropericardium syndrome and gizzard erosion) [9]. In contrast, FAdV-B strains were detected from various pathologies (arthritis, enteritis, inclusion body hepatitis and hemorrhages, among others), but no primary disease was associated with them [10,11,12,13]. The FAdV-5 prototype strain 340 was isolated decades ago during a routine survey carried out in hens without specific pathology [7].
Our recent results demarcated a proposed new genotype within the species: strain 40440-M/2015 Debrecen (hereafter strain 40440) shared lower sequence identity with reference strain 340, in the relevant antigenic regions, than the closest related known FAdV serotypes shared with each other [12]. Thus, we wanted to investigate if this newly recognized level of genetic diversity in FAdV-B has consequences on serum neutralization, and to scrutinize the possibility of a novel serotype within the species. Furthermore, our results shed new light on the actual diversity of FAdV-B, whose molecular evolution has remained with many gaps due to the lack of priority given to this fowl aviadenovirus species in regard to clinical manifestations.
2. Materials and Methods
2.1. FAdV-B Isolates
All FAdV-B strains included in this study were isolated from commercial chickens from different European countries during the years 2015–2018. The origin of the strains, along with the clinical documentation for the corresponding flock, if available, are summarized in Table 1.
The complete genome of strain 40440 (MG953201) has been reported previously [12]. The remaining fourteen strains were obtained from samples submitted for routine diagnostics to the Clinic for Poultry and Fish Medicine, University of Veterinary Medicine, Vienna, Austria. The presence of FAdV-B strains in these samples was determined, after propagation on chicken embryo liver (CEL) cells, by amplification and sequencing of the hexon loop-1 (L1) gene region [14]. Prior to further processing, all isolates were plaque purified 3 times. For this, 0.1 mL of serial tenfold virus dilutions were inoculated on a CEL monolayer grown in 6-well plates, which were afterwards incubated with 3 mL tissue culture medium. After 24 h, the medium was replaced by 3 mL M199 tissue culture medium containing 1.26% agarose 4% (Thermo Fisher, Vienna, Austria). Upon visible formation of plaques, individual plaques were excised from the agarose layer using a 1000 μL pipette tip with an orifice cut with a sterile scissor to fit the plaque. The plaque was transferred to an Eppendorf tube and dissolved in 1–2 mL medium, to serve as inoculum for successive rounds of plaque purification.
2.2. Sequencing and Genome Assembly
Whole-genome sequencing of the 14 strains was performed on an Illumina platform (MiSeq V3, Central Service Facility NGS Unit, Vienna, Austria). Paired-end libraries were generated, and samples were multiplexed in a single lane, separated by barcoding. Contaminating chicken genome reads were excluded by mapping them against the Gallus gallus genome (NCBI Genome GRCg6a, RefSeq assembly GCF_000002315.6) using MIRA 4.0.2 [15]. The resulting reads were error corrected and normalized to a 40-times coverage using BBNorm from the BBTools suite. The normalized reads were quality trimmed, and then mapped to the genome of reference strain 340 (KC493646) using the Geneious mapper at the highest sensitivity, and allowing it to reveal structural variants, insertions and deletions of any size [16]. For the resulting consensus sequences, the same reads were mapped using the same sensitivity settings to obtain the final consensus sequences. Genome annotations were transferred from strain 40440 using Geneious and manually checked and edited. The exported GenBank flat files were converted to five-column, tab-delimited feature tables using the GB2sequin for submission purposes [17]. The complete genome sequences were deposited in the GenBank, under accession numbers OK283042-OK283055, whereas sequence reads were archived under the NCBI BioProject, under the accession number PRJNA781911.
2.3. Phylogenetic Analysis
Phylogeny was inferred using pairwise sequence identity analyses and phylogenetic tree reconstructions.
Pairwise sequence identities were measured using MAFFT for alignments in the Sequence Demarcation Tool [18]. Sequence identities were calculated based on the complete genomes and on the amino acid (aa) sequences of: the complete DNA polymerase, penton base, hexon and fiber, the hexon hypervariable loop-1, and the fiber knob, as well as the concatenate of the major capsid proteins’ serodeterminant regions (penton base, hexon loop-1, hexon loop-2 and fiber knob).

Table 1.
FAdV-B isolates investigated in this study.
Table 1.
FAdV-B isolates investigated in this study.
| Strain Name (GenBank acc.no.) | Country, Year of Origin | Type of Sample | Clinical Signs, Pathologies | Other Bacterial/Viral Findings |
|---|---|---|---|---|
| 14-24408 (OK283042) | Austria a, 2014 | Organ pool (liver, intestine, caecal tonsils) | Birds dead on farm | Viral: none |
| 15-368 (OK283043) | Austria b, 2015 | Organ pool (liver, intestine, caecal tonsils) | Broilers, dead on farm | Viral: none |
| 15-1401 (OK283044) | Austria c, 2015 | Organ pool (liver, intestine, caecal tonsils) | Birds dead on farm | Viral: none |
| 15-3466 (OK283045) | Austria d, 2015 | Organ pool (liver, intestine, caecal tonsils) | Broilers, dead on farm No lesions recorded | FAdV-1 |
| 15-4225 (OK283051) | France, 2015 | Pancreas | 21-day-old chickens with severe tenosynovitis (2–3% incidence) | Tendons: Reovirus |
| 15-4616 (OK283046) | Austria e, 2015 | Organ pool (liver, intestine, caecal tonsils) | Broilers, dead on farm Liver swollen, perihepatitis Pericarditis Ascites | E. coli, Staphylococcus spp. Reovirus |
| 15-6270 (OK283047) | Austria f, 2015 | Organ pool (liver, intestine, caecal tonsils) | Broilers, dead on farm Liver swollen, necroses Ascites Intestinal dilatation, foamy contents | Proteus spp. in all organs |
| 15-6541 (OK283048) | Austria g, 2015 | Organ pool (liver, intestine, caecal tonsils) | Broilers, dead on farm | E. coli, Staphylococcus spp. |
| 17-25702 (OK283049) | Hungary a, 2017 | Caecal tonsils | Broiler breeders, dead on farm | Viral: none |
| 18-907 (OK283050) | Hungary b, 2018 | Caecal tonsils | Broiler breeders, dead on farm | Viral: none |
| 18-6238 (OK283052) | Germany, 2018 | Cell culture supernatant (material of origin unknown) | n.r. | n.c. |
| 18-6239 (OK283053) | ||||
| 18-11753 (OK283054) | Hungary a, 2018 | Caecal tonsils | Broiler breeders, dead on farm | n.c. |
| 19-7207 (OK283055) | Hungary c, 2018 | Cloacal swab pool | n.r. | n.c. |
a–g Samples from the same country, but separate farms in different locations, are indicated by different superscript letters. n.r.—not reported; n.c.—not conducted.
The strains were compared phylogenetically to FAdV-5 reference strain 340 based on tree inferences on the complete genomes and complete hexon aa sequences. Furthermore, a phylogenetic tree was inferred using all FAdV-B (NCBI: txid190062) hexon loop-1 nucleotide sequences. Here, misannotated and low-quality sequences were excluded from the analysis after a preliminary affirmatory molecular typing and quality check. Misannotation is, unfortunately, not limited to FAdV-B sequence entries; therefore, the strain 340 hexon loop-1 nucleotide sequence was used as a query in an NCBI BLASTn search to find further virus strains not listed in NCBI Taxonomy ID 190062 (species FAdV-B). As entry MK757473 (erroneously designated as fowl aviadenovirus 5 isolate LYG, BLASTX score: 494) was found to not represent a FAdV-B strain, a BLASTX score threshold >494 was applied. Analyzed strains are summarized in Table 2. Sequence alignments were conducted using MAFFT: the E-INS-i algorithm was used for the genomes and the G-INS-i for both hexon alignments [19]. The alignments were edited using Gblocks. Evolutionary models were predicted using ModelTest-NG: the GTR + I + G model was selected for the genomes and the partial hexons, whereas the JTT + I + G was selected for the complete hexons [20]. Phylogenetic trees were reconstructed using RAxML-NG; the robustness of the trees was tested with a non-parametric bootstrap calculation using 1000 repeats. Phylogenetic trees were visualized using MEGA 7 [21], and bootstrap values were given as percentages if they reached ≥75%.
2.4. Cross-Neutralization Testing
Cross-neutralization testing was performed in order to examine the serological relationships among the viruses, as well as to assess the possible effects of sequence differences on mutual recognition between current field isolates of this study and strain 340, the FAdV-5 (FAdV-B) reference strain, isolated in Northern Ireland around 1970 [7]. Field strains included were 40440 and three representatives (15-6541, 15-4225, and 18-6238) from the remaining FAdV-B strains. The latter three were selected based on the complete genome phylogenetic analysis to represent relatively distant strains; however, all three selected strains shared complete aa sequence identity in the major capsid proteins among each other, while featuring few aa differences compared to 40440 and 340.
Monospecific antisera were raised against each strain in specific pathogen-free chickens (VALO BioMedica GmbH, Osterholz-Scharmbeck, Germany) immunized intramuscularly with a mixture of inactivated (1% formaldehyde) virus and adjuvant (GERBU LQ no. 3000, GERBU Biotechnik GmbH, Heidelberg, Germany). The procedures used on the experimental birds were discussed and approved by the institutional ethics committee and the national authority according to §26 of the Law for Animal Experiments, Tierversuchsgesetz 2012–TVG 2012 (license numbers GZ 68.205/0158-WF/V/3b/2014, GZ 68.205/0044-WF/V/3b/2016).
Neutralizing antibody (nAb) titers of the freshly prepared sera were pre-determined in a microtiter assay on CEL cells, testing each serum in duplicate against its corresponding (homologous) virus. Heat inactivated, serially diluted (log23–log214) sera were incubated with 100 TCID50 of the virus. After five days of incubation at 37 °C in 5% CO2, the wells were checked for cytopathic effect (CPE) using light microscopy. For the subsequent cross-neutralization assay, the sera were standardized to a concentration of 20 serum units against the homologous strain, diluted at a ratio of 1:20, then tested in serial two-fold dilutions against all viruses under the conditions outlined above [22].
An 8-fold difference in the titer compared to the homologous value was assumed for serotype differentiation [7,23].
Table 2.
Fowl aviadenovirus B strains, phylogenetically analyzed based on their hexon loop-1 nucleotide sequences. The accession numbers of the newly sequenced strains are given in bold.
Table 2.
Fowl aviadenovirus B strains, phylogenetically analyzed based on their hexon loop-1 nucleotide sequences. The accession numbers of the newly sequenced strains are given in bold.
| Accession Number | Strain Identifier | Country of Origin | Year of Origin | Taxonomic Lineage (If not FAdV-B) |
|---|---|---|---|---|
| AF508953 | TR22 | Japan | 1960s | |
| KC493646 | 340 | Northern Ireland | 1970s | |
| EF442425 | Rostov/2007/02/chicken/B | Russia | 2007 | Aviadenovirus |
| FN869989 | 08-21472 | Austria | 2008 | |
| FN869990 | 08-8669 | Austria | 2008 | |
| FN869987 | 09-7470-2 | Hungary | 2009 | |
| FN869988 | 09-7473-2 | Hungary | 2009 | |
| FN869991 | 09-6893 | Hungary | 2009 | |
| HQ697592 | K318/09 | South Korea | 2009 | Fowl aviadenovirus D |
| JF304111 | 8844 | Hungary | 2010 | |
| KC750798 | 160 | Hungary | 2011 | |
| KC750799 | 177 | Hungary | 2011 | |
| KP274034 | FAdVB_CGOU224 | Cote d’Ivoire | 2012 | unclassified aviadenovirus |
| KP274035 | FAdVB_CPON047 | Cote d’Ivoire | 2012 | unclassified aviadenovirus |
| KP274036 | FAdVB_CPAU286 | Cote d’Ivoire | 2012 | unclassified aviadenovirus |
| KP274037 | FAdVB_CPON040 | Cote d’Ivoire | 2012 | unclassified aviadenovirus |
| KP274038 | FAdVE_CDAO182 | Cote d’Ivoire | 2012 | unclassified aviadenovirus |
| KP828383 | GB 1643 | Germany | 2012 | |
| MG953228 | 9892 | Hungary | 2013 | |
| MT500572 | D2453 | Ukraine | 2013 | |
| MG953211 | 2255 | Hungary | 2014 | |
| OK283042 | 14/24408 | Austria | 2014 | |
| MG953201 | 40440 | Hungary | 2015 | |
| MG953219 | 45871 | Hungary | 2015 | |
| MG953222 | 5626 | Hungary | 2015 | |
| MG953223 | 70147 | Hungary | 2015 | |
| OK283043 | 15/368 | Austria | 2015 | |
| OK283044 | 15/1401 | Austria | 2015 | |
| OK283045 | 15/3466 | Austria | 2015 | |
| OK283046 | 15/4616 | Austria | 2015 | |
| OK283047 | 15/6270 | Austria | 2015 | |
| OK283048 | 15/6541 | Austria | 2015 | |
| OK283051 | 15/4225 | France | 2015 | |
| MK509019 | NGR_FAdV_Ch2 | Nigeria | 2017 | Fowl aviadenovirus C |
| OK283049 | 17/25702 | Hungary | 2017 | |
| OK283050 | 18/907 | Hungary | 2018 | |
| OK283052 | 18/6238 | Germany | 2018 | |
| OK283053 | 18/6239 | Germany | 2018 | |
| OK283054 | 18/11753 | Hungary | 2018 | |
| OK283055 | 19/7209 | Hungary | 2019 | |
| AF339916 | ATCC VR-828, VR-1854, IBH-2A | n.a. | n.a. | Fowl aviadenovirus D |
| AF508953 | TR22 | Japan | 1960s | |
| KC493646 | 340 | Northern Ireland | 1970s | |
| EF442425 | Rostov/2007/02/chicken/B | Russia | 2007 | Aviadenovirus |
| FN869989 | 08-21472 | Austria | 2008 | |
| FN869990 | 08-8669 | Austria | 2008 |
n.a.—not available.
2.5. Modelling of the Major Capsid Proteins
In spite of a notable genetic distance, in other words, a relatively low sequence identity, the divergent FAdV-B strains did not represent a novel serotype. Therefore, to determine the structural background of this, the three-dimensional protein models of the serodeterminant major capsid proteins were generated for the reference (340) and the divergent FAdV-5 strain (40440), as well as an FAdV-8a strain (TR59), based on the following aa sequences: AGL34683, QCC26485 and ANJ02525 (penton base); AGL34687, QCC26479 and ANJ02529 (hexon); and finally AGL34695, QCC26484 and ANJ02537 (fiber). FAdV-8a served as a positive control, as it represented a clearly distinct serotype compared to any FAdV-5 strain. All models were generated using the Robetta structure prediction server [24]. Not only did this application generate the monomer models, but it also created the appropriate biological assemblies: pentamer from penton base, and trimers from hexon and fiber subunits. The output protein model structures were refined with the MacroModel energy minimization module of the Schrödinger Suite [25] to eliminate steric conflicts between the side-chain atoms. The models were visualized in VMD version 1.9.3 [26]. Electrostatic potential maps were calculated with Adaptive Poisson–Boltzmann Solver (APBS) version 1.3 using the linearized Poisson–Boltzmann method [27] with a dielectric constant of 78.0 and 2 for the water solvent and the protein core, respectively. The partial charges in the electrostatic potential were calculated with PDB2PQR [27].
3. Results
3.1. Propagation of FAdV-B Strains
Five days after the infection of CEL cells, every FAdV-B strain developed a visible CPE including plaque formation, rounding and condensation of cells, and detachment from the plate, similar to those caused by the reference strain 340. No major differences were recorded in the morphology and/or the progression of CPE between investigated strains.
Five strains’ infective titers were determined in the course of cross-neutralization testing (see Section 3.4); these were 104.4 TCID50/mL for strain 340, 106.7 TCID50/mL for strain 40440 and 18-6238, 108.2 TCID50/mL for strain 15-4225, and 109 TCID50/mL for strain 15-6541.
3.2. Genome Sequences
The complete genome sequences of the 14 FAdV-B strains were determined. The genome lengths varied between 45.741 and 45.838 bp, with a G + C content of 56.5–56.6%. After the final sequence read mapping, the final read coverage minimums were between 5 and 11, and the means were between 76.4 and 80.3. The same open reading frame content and orientation of strain 40440 was observed in every new FAdV-B genome with 36 open reading frames, inverted terminal repeats of 86 bp, and two tandem repeat regions; the first one was located in the intergenic regions between ORF27 and GAM-1, and the second between GAM-1 and ORF17.
3.3. Phylogenetic Analysis
All serodeterminant surface antigens shared 100% aa sequence identity among the newly sequenced strains; the penton base and the hexon genes shared 100% nucleic acid identity too, whereas the fiber genes had 0–2 nucleotides in difference. Additionally, the penton base, hexon and fiber knob of strain 40440 shared 100% aa identity with the analyzed strains’ corresponding proteins. The same proteins shared a lower sequence identity (89.80–99.63%) with those of strain 340. In the DNA polymerase aa sequences of the newly sequenced strains, 0–2 differences were counted. The sequence identity values are summarized in Table 3, and the alignment of the hexon loop-1 is available in Figure 1.
Table 3.
Pairwise sequence identity values of Fowl aviadenovirus B strains.

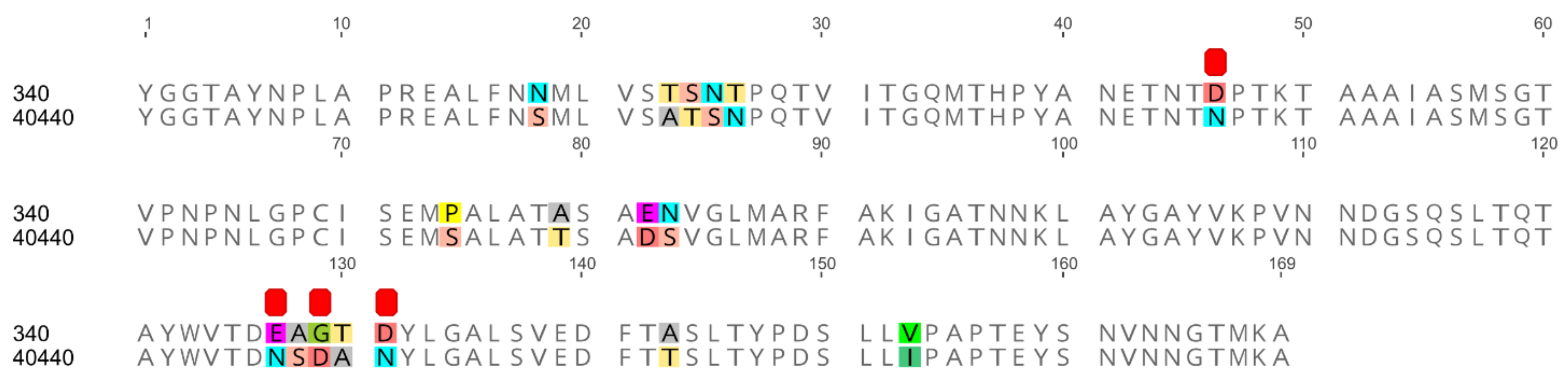
Figure 1.
Amino acid alignment of hexon loop-1 sequences originating from two fowl adenovirus 5 strains: the reference strain 340 and another one (40440). The newly sequenced divergent strains are identical on the amino acid level to strain 40440 on this stretch. Differences are highlighted; furthermore, amino acid substitutions affecting the electrostatic charge of the protein are denoted using red squares.
Based on the complete genomes’ phylogenetic tree reconstruction, all newly sequenced strains were placed on a monophyletic branch, and strain 40440 was most closely related to the recently described recombinant D2453 [13]. Analyzing the hexon aa sequences, it was confirmed again that the newly analyzed FAdV-B strains share an identical hexon protein with strain 40440. Furthermore, the analysis of the hexon loop-1 nucleotide sequences clustered almost all FAdV-B strains into three clades, members of which shared almost identical loop-1 nucleotide sequences. Only strain TR22 constituted an independent branch, that was not monophyletic with other FAdV-B members; furthermore the distance of TR22 from other FAdV-B strains was similar to the observable interspecies distances. The phylogenetic tree reconstructions are shown in Figure 2.
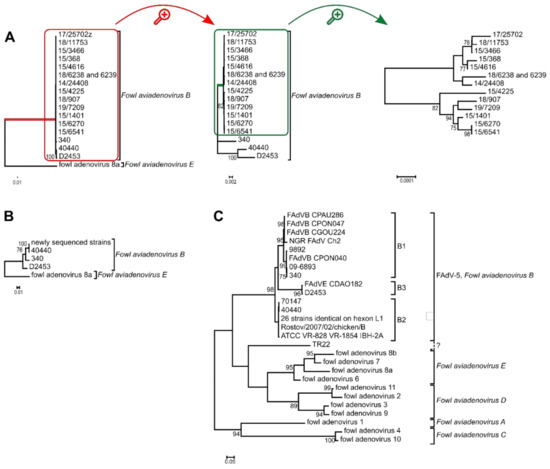
Figure 2.
Phylogenetic analysis of Fowl aviadenovirus B strains based on the (A) complete genome sequence, (B) the hexon amino acid sequence and the (C) hexon loop-1 (L1) nucleotide sequence. Trees (A,B) were rooted on fowl adenovirus 8a, whereas tree (C) was midpoint rooted. (A) Branch lengths were short within the species Fowl aviadenovirus B; thus, the red highlighted branch of the species is shown separately. From this tree, the green highlighted branch of the newly sequenced strains was separated again to reveal branch lengths among these strains too. (C) The 26 identical strains: 160, 177, 2255, 5626, 8844, 45871, 08-21472, 08-8669, 09-7470-2, 09-7473-2, 14/24408, 15/1401, 15/3466, 15/368, 15/4616, 15/6270, 15/6541, 17/25702, 18/11753, 15/4225, 18/6238, 18/6239, 18/907, 19/7209, GB 1643, K318/09. See Table 2 for further details.
3.4. Neutralizing Antibodies and Cross-Neutralization of FAdV-B Strains
Neutralizing titers, determined in sera collected at 28, 35 and 42 days post immunization, were in the upper range of the routinely tested interval for FAdV-5 reference strain 340 (log211–log212), and field strain 18-6238 (log212–log213). Since no end point could be determined within this range for any of the remaining antisera, additional doubling serum dilutions up to log220 were included. This revealed titers of log214–log215 for 40440 antisera, while injections with strains 15-4225 and 15-6541 revealed antisera reaching log216–log218 and log218–log219, respectively. A small but consecutive rise in titer between the first and last time point was recorded in each tested individual, indicating that nAb production was ongoing 4–6 weeks after the antigen injection.
In the cross-neutralization assay, mutual recognition occurred between all contemporary FAdV-B field strains, including the genetically most distant 40440 (Table 4). An identical level of neutralization was shown between the three newly sequenced field strains, and an up to two-fold lower neutralization compared to the respective homologous titer of 40440. Antisera against all four current field strains were also able to neutralize reference virus 340, with a two- to four-fold titer reduction compared to the homologous reaction. However, all of the reactions with 340 were unanimously one-directional, as the antiserum against 340 exhibited no neutralizing activity against any of the current FAdV-B field isolates.
Table 4.
Cross-neutralization testing between selected FAdV-B field isolates and the historical reference strain 340. The extent of neutralization is expressed as the reciprocal titer of antisera, standardized to 20 serum units against 100 TCID50 of the homologous virus. Values highlighted in grey represent the homologous neutralization and the reference for all other reactions in the respective vertical column.
3.5. Modelling of the Major Capsid Proteins
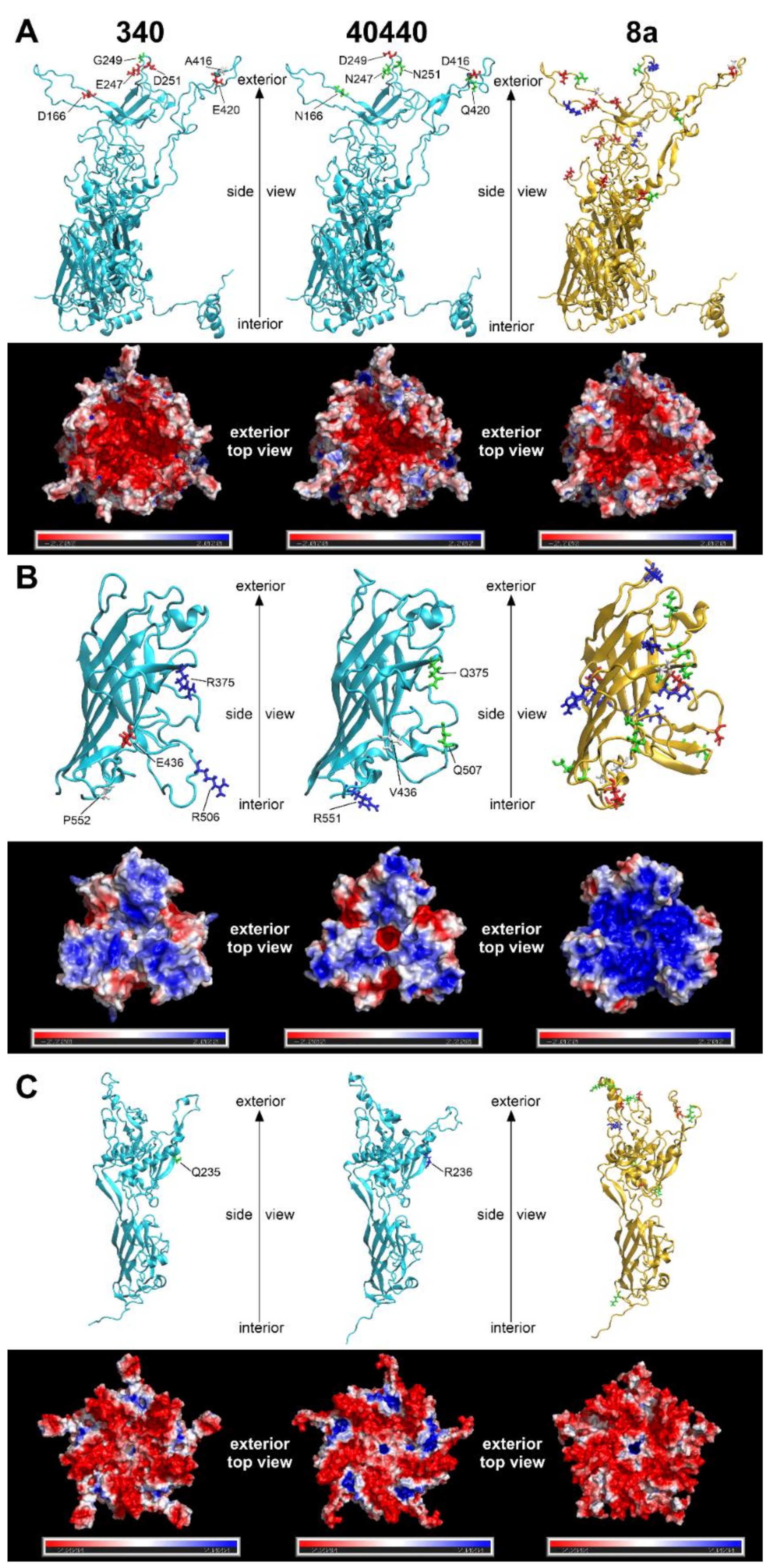
The predicted structures of capsid proteins are presented in Figure 3. On the hexon trimers, strain 40440 had a positively charged surface potential patch in its peripheral region; this patch was smaller and less positively charged in FAdV-8a, and was neutral in strain 340. The central, negatively charged depression was most extensive in strain 340, whereas it was similar in size in strain 40440 and FAdV-8a. On the fiber trimers, a negatively charged peripheral indentation was present in both FAdV-5 strains, though it was more prominent in strain 40440. In the latter one, a small, marked negatively charged region was observed around the central cavity, too, whereas the FAdV-8a fiber head was predominantly strongly positively charged almost on its entire surface. On the penton pentamer, positively charged dents were observed in FAdV-5 strains, which were charged more strongly in strain 40440. The dents were observable in FAdV-8a, too, but were uncharged. A positively charged small central cavity was observed both in strain 40440 and in FAdV-8a; the surrounding region was slightly negative in strain 40440 and more neutral in FAdV-8a.
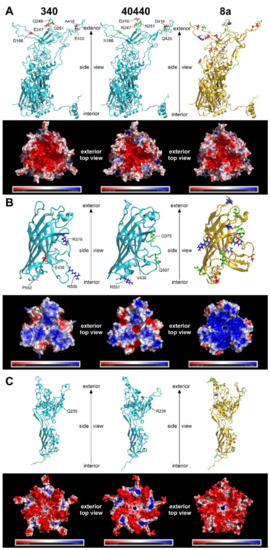
Figure 3.
Three-dimensional major capsid protein models of strains 340 and 40440 (both serotype fowl adenovirus 5 [FAdV-5]) and FAdV-8a: (A) complete hexon; (B) fiber knob (amino acid stretches for strain 340: 349–553, 40440: 349–554, FAdV-8a: 314–524); and (C) penton base (amino acid stretches for strain 340: 76–542, 40440: 77–543, FAdV-8a: 81–552). The upper strips of each subset show the monomer subunits in cartoon representation; here, amino acid substitutions affecting the electrostatic charge of the protein are highlighted. For strains 340 and 40440, substitutions compared to each other, and for fowl adenovirus 8a, substitutions compared to strain 340, are highlighted. Red and blue are charged (negatively and positively, respectively), green are polar, and white are nonpolar amino acids. The lower strips of each subset represent the electrostatic surface potentials of the trimeric (hexon and fiber knob) or pentameric (penton base) proteins, where red color represents regions with a potential value below −2.0 kT, white represents 0.0 kT, and blue represents regions above +2.0 kT.
Differences in the total quantity of charged aas were also compared between strains 340 and 40440 based on Figure 3. On a hexon monomer, four negatively charged aas were counted on strain 340, and two on strain 40440. On a fiber monomer, two positively charged and a negatively charged aa were counted on strain 340, and a single positively charged aa was noted on strain 40440. Finally, on a penton base monomer, a single positive aa was noted on strain 40440. This analysis compared only the total quantity of the charged aas, but all aa changes were scrutinized, focusing on charge alterations based on sequence alignments. These amino acid changes of the major capsid proteins are summarized in Table 5.
Table 5.
Amino acid substitutions of the major capsid proteins.
4. Discussion
In previous studies, we had proposed the demarcation of a second genotype for genetically divergent strains within species FAdV-B [10,11], based on the finding that the genetic distances between the divergent and the official FAdV-5 reference strains had been higher on every analyzed genomic stretch than those measured between the closest related, acknowledged serotype [12]. These results included all adenoviral serodeterminant surface antigens and a concatenate of these; thus, it was assumed that such a genetic distance predicts a novel serotype, demarcated by serum neutralization. By definition, a serotype either exhibits no cross-reaction with others, or shows a homologous/heterologous titer ratio of greater than 8 (in both directions) [7,23]. Four contemporary divergent FAdV-B strains—sharing identical sequences in the major adenoviral serodeterminant regions—were tested in this study, together with reference strain 340 in a virus cross-neutralization assay. In this setting the historical prototype 340 was singularized from the group of contemporary strains by the fact that its respective antiserum completely failed to neutralize any of the contemporary strains; these, less surprisingly, shared full recognition among each other underlining their close serological relationship. On the other hand, the homologous/heterologous titer ratios of antisera against the divergent strains were below the minimal threshold value of 8; this, by definition, contests a presumptive new serotype within species FAdV-B.
The major capsid proteins of adenoviruses are the hexon, the penton base and the fiber [2], and the external surface of these, mainly the hypervariable loops of the hexon protein and the fiber knob [28,29], act as antigenic determinants [23,30,31,32]. Genetic changes in these surface proteins are a predictor for serological distancing [10,28]; thus, the major capsid proteins were modelled to elucidate why the serodeterminants—despite genetic distance—did not result in a different neutralization phenotype in the actual case. Due to its numerical abundance in the capsid [33], the hexon loop-1 is the dominant component of the serotyping response [34,35]. Although 26 aas were substituted in the divergent strains’ complete hexon compared to that of strain 340 (Figure 1), none of these reversed the charge of the residue in question, and only one caused a nonpolar-to-charged aa change. A further five substitutions altered the charge of the residue only slightly by a change from a polar acidic aa to a still-polar but uncharged one, or vice versa. Similarly, charge reversal was not observed in any residue of the fiber knob or the penton base; only nonpolar-to-charged or polar-to-charged changes were observed. Conversely, in a clearly distinct serotype, serotype FAdV-8a, a higher number of marked charge alterations (Table 5) and an apparently different fiber knob electrostatic surface potential (Figure 3) were observed. After analyzing the substitutions and the resulting electrostatic surface potentials, we conclude that genetic distance alone might be insufficient as a criterion for serotype inference of fowl adenoviruses. Consequently, to acquire a more detailed understanding of the molecular basis for neutralization phenotypes, comparative bioinformatics approaches—taking into account the location of changed residues within the molecules´ tertiary structure and corresponding changes of the electrostatic surface potentials—should also be included, alongside sequence divergence in future studies.
The unidirectional recognition between the strains indicates (Table 4), however, that the similarity of all serodeterminant components was, in summary, still enough to enable at least a one-way cross-neutralization. On the other hand, the observed amino acid changes in the capsid proteins (Figure 1 and Figure 2B, Table 3 and Table 5) cause the divergent strains to be on the borderline of representing a new serotype. Although not a new serotype, all divergent strains, according to their neutralization behavior, represent prime strains of FAdV-B. A prime strain induces antibodies that neutralize the originally recognized prototype strain, while it is, in turn, poorly (or not) neutralized by the prototype antiserum. This phenomenon has been described already among species FAdV-D and -E strains [35]. To investigate the possibility that only one of the components, and its cognate antibody fraction, was responsible for heterologous recognition, and that a lack thereof caused one-sided failure to neutralize (i.e., antiserum to strain 340 vs. the divergent viruses), we screened the antibody contents of all of the tested sera using Western blots with whole virus preparations. Although Western blotting might also detect antibodies against non-neutralizing epitopes the blots still indicated the presence of the full capsid-directed antibody repertoire in all of the sera (data not shown). This makes it less likely that the antiserum to strain 340 was devoid of a specific antibody fraction, and that this is an explanation for accidentally-assigned prime strains. Amino acid differences in the hexon might well contribute to the noticed one-way neutralization (Figure 1). In addition, the complete lack of neutralization of the closely related strains by serum raised against reference strain 340 argues for additional mechanisms. Thus, we turned, again, to the conformational and surface potential models.
One of the conditions required for basic protein–protein interactions is electrostatic complementarity. In our case, the hexon of strain 340 carried exactly twice as many negative charges (Figure 3A) in the antigenic L1 and L2 regions as strain 40440, which is the prime strain. Thus, antibodies, raised against the reference strain’s hexon have twice as many positive charges as those against the prime strain hexon. Simultaneously, the hexon of the prime strain bears significant positively charged surface regions that are likely to repel the more positive antibodies raised against the reference strain, so no neutralization occurs. Conversely, antibodies raised against the hexon of a prime strain with half the number of positive charges are capable of binding to the less positively charged hexon of the reference strain, leading to successful neutralization. The exact description of cross-neutralization is further complicated by the fact that, in addition to the hexon, two other proteins—the fiber knob and penton base—also have antigenic regions. In the present case, however, the antigenic regions of the fiber knob and penton base of the reference strain are similar to those of the prime strain, so these proteins probably do not contribute to this prime strain phenomenon. Finally, it needs to be kept in mind that antibodies against different capsid proteins have synergistic potential with regard to neutralizing activity [36].
Essentially identical antigenic sets with the serodeterminants of 40440 were found in all of the newly sequenced viruses, though they spanned a period of four years and a sampling region of four countries. Although other variants had been detected earlier [10,11,37], this serodeterminant set was so prevalent in our study that we speculate that it represents the currently dominating wild type having replaced the original prototype. Unfortunately, FAdV-B is by far the most underrepresented of all of the species, with regard to its documentation throughout the decades, from the isolation of strain 340 to recent strains; this is mainly due to a lack of clinical relevance attributed to those strains. However, the finding that even the highly conserved, slowly evolving DNA polymerase gene showed greater variability among the strains than the surface antigens suggests that this serodeterminant set was acquired long before its recent detection [38,39]. Furthermore, the analysis of all of the available FAdV-B hexon loop-1 sequences (Figure 2C) revealed that 75% of the strains clustered into this genotype, genotype B2 [10]. It is tempting to hypothesize that this antigenic set might give an evolutionary advantage to the strains, and we can observe a shift in the serodeterminants of FAdV-5. The new strains were already not neutralized by the antiserum of the reference strain; on the other hand, any sera against the new, divergent strains (genotype B2) neutralized the reference strain (genotype B1). This alone already provides an evolutionary advantage for the divergent strains; these strains are possibly capable of infecting chickens already seroconverted against the reference strain, whereas the opposite is unlikely true with the presence of neutralizing antibodies. Such a hypothesis of antigenic shift should be handled cautiously, as the average life expectancy of a broiler chicken is only 5–7 weeks. Although not impossible, it is not probable that two adenovirus infections will occur during this short period; thus there would be no evolutionary pressure for such a shift. On the other hand, breeders or layer pullets live much longer, so there is a theoretical possibility for such an explanation. Concurrent infections, with more than one adenovirus type, do occur in chickens just as in humans [11,14,40,41,42,43,44,45,46,47]; this is now strengthened in our own dataset by the example of strain 15-3466, which was co-isolated with a FAdV-1 strain.
The analysis of all FAdV-B strains (Figure 2C) indicated, again, the questionable typing result of strain TR22. This strain was isolated in Japan in the 1960s and was serotyped as FAdV-5 [48]. However, our findings confirmed previous results [49], as the strain was not monophyletic with other FAdV-B strains. The serological typing results of this strain remain questionable, and a repeated cross-neutralization assay would be supported.
TR22 was isolated in Japan, and strain 340 in Northern Ireland, but the majority of more recent FAdV-B isolations or detections happened in two geographical regions: Central Europe and West Africa. Since 2007, 31 FAdV-B strains (Table 2) have originated from France (one), Germany (three), Austria (nine), Hungary (sixteen), Ukraine (one) and Russia (one), where the latter strain was isolated in Rostov, about 100 km from the Ukrainian border [10,11,12,13,50,51]. A further two strains (NCBI Nucleotide: MT525091, MW353019), originating from Poland, were excluded from the analysis based on low sequence quality [52]. Moreover, six strains were detected in West Africa: in Cote d’Ivoire (five) and in Ibadan, Southwest Nigeria (one) [53]. Thus, the only geographical exception since 2007 is a single strain from South Korea (HQ697592) [54], although numerous typing results have recently been published worldwide [40,41,42,43,44,45,55,56,57,58,59,60,61,62,63]. Seemingly, species FAdV-B has been limited almost exclusively to Central Europe and West Africa in recent years.
The exact cause of this is not known. It might be hypothesized that FAdV-B strains have adapted evolutionarily to these geographical regions or husbandry technologies, and have persisted there, whereas this species was superseded by other FAdV species in other regions; for example, the trade of hatching eggs is widely practiced in Europe, and enables the spread of a certain serotype due to vertical transmission. Another possibility is the exact contrary of the former: a recent spread of FAdV-B strains in these locations. This might be supported by the emergence of two genotypes within the species: B2, and a novel one, B3. As mentioned before, B2 strains represent 75% of all FAdV-B strains, and a further two strains (isolated in 2012 and 2013) constitute a relatively independent branch, characterized by a long branch length; thus the genotype B3 was established (Figure 2C). This analysis was based on the hexon loop-1 nucleotide sequence, a genomic stretch under the highest evolutionary pressure of the host’s immune system [35]. Other parts of the genome are far from this mutation rate, and intraspecies distances compared on the complete genome (Figure 2A) or the DNA polymerase (Table 3) are minuscule.
The members of almost every FAdV species are associated with specific diseases. Namely, FAdV-A (serotype FAdV-1) with gizzard erosion, FAdV-C (FAdV-4) with hepatitis–hydropericardium syndrome, and FAdV-D (FAdV-2, -11) and FAdV-E (FAdV-8a, -8b) with inclusion body hepatitis [5]. In this respect, species FAdV-B is an exception, since such strains have not been found to be the aetiological agent of a well-circumscribed disease. On the contrary, FAdV-B strains have already been found in connection with tenosynovitis, inclusion body hepatitis, airsacculitis, pericarditis, enteritis, cardiac decompensation or nephrosis in chickens, and have also been isolated from healthy mallards [10,11,12,50,64]. No clear pattern was observed in our study either; FAdV-B was mainly an incidental finding, and lesions were attributed to other viral/bacterial infections diagnosed in the flocks. Strains belonging to this species are most likely facultative pathogens of chickens, and additional immunosuppressive effects are necessary for disease manifestation.
The genome sequences of fourteen FAdV-B strains, representing currently circulating strains in Europe, were determined, and four selected strains were compared using serum cross-neutralization to reference strain 340. Based on previous phylogenetic analyses, it was hypothesized that the divergent FAdV-B strains might represent a novel serotype within the species, but the actual cross-neutralization tests refuted this. This research underlines the importance of traditional neutralization assays, optimally performed as reciprocal testing when diagnostic difficulties are suspected due to prime relationships. Furthermore, we provide a snapshot of the current diversity of the species Fowl aviadenovirus B, for which a long-term gap in epidemiological documentation has obviously lead to the widely unrecognized emergence and spread of particular antigenic variants.
Author Contributions
Conceptualization, G.L.K. and A.S.; methodology, G.L.K. and A.S.; validation, M.H.; formal analysis, G.L.K. and A.S.; investigation, A.S. and Á.G.; resources, M.H.; data curation, G.L.K. and Á.G.; writing—original draft preparation, G.L.K. and A.S.; writing—review and editing, M.H.; visualization, G.L.K. and Á.G.; supervision, M.H.; project administration, M.H.; funding acquisition, M.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was partially funded by the National Research, Development and Innovation Office (grant number NN140356) and partially implemented with the support provided by the Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund, financed under the TKP2021-EGA-01 funding scheme of the National Research, Development and Innovation Office. The research of GLK is supported by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences.
Institutional Review Board Statement
The procedures used on the experimental birds were discussed and approved by the institutional ethics committee and the national authority according to §26 of the Law for Animal Experiments, Tierversuchsgesetz 2012–TVG 2012 (license numbers GZ 68.205/0158-WF/V/3b/2014, GZ 68.205/0044-WF/V/3b/2016).
Informed Consent Statement
Not applicable.
Data Availability Statement
The complete genome sequences were deposited to the GenBank, under accession numbers OK283042-OK283055, whereas sequence reads were archived under the NCBI BioProject accession number PRJNA781911.
Acknowledgments
Phylogenetic calculations were performed using computing resources provided by KIFÜ, Hungary.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Harrach, B.; Tarján, Z.L.; Benkő, M. Adenoviruses across the animal kingdom: A walk in the zoo. FEBS Lett. 2019, 593, 3660–3673. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, J.; Pérez-Illana, M.; Martín-González, N.; San Martín, C. Adenovirus Structure: What Is New? Int. J. Mol. Sci. 2021, 22, 5240. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.; Dolja, V.; Krupovic, M.; Varsani, A.; Wolf, Y.; Yutin, N.; Zerbin, I.M.; Kuhn, J. Create a Megataxonomic Framework, Filling All Principal Taxonomic Ranks, for DNA viruses Encoding Vertical Jelly Roll-Type Major Capsid Proteins. Available online: https://talk.ictvonline.org/ictv/proposals/2019.003G.zip (accessed on 10 June 2021).
- Harrach, B.; Kaján, G.L. Aviadenovirus. In The Springer Index of Viruses; Tidona, C.A., Darai, G., Eds.; Springer: New York, NY, USA, 2011; pp. 13–28. ISBN 978-0-387-95918-4. [Google Scholar]
- Schachner, A.; Matos, M.; Grafl, B.; Hess, M. Fowl adenovirus-induced diseases and strategies for their control—A review on the current global situation. Avian Pathol. 2018, 47, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Kiss, I.; Homonnay, Z.G.; Mató, T.; Bányai, K.; Palya, V. Research Note: An overview on distribution of fowl adenoviruses. Poult. Sci. 2021, 100, 101052. [Google Scholar] [CrossRef] [PubMed]
- McFerran, J.B.; Clarke, J.K.; Connor, T.J. Serological classification of avian adenoviruses. Arch. Gesamte Virusforsch 1972, 39, 132–139. [Google Scholar] [CrossRef]
- Marek, A.; Kosiol, C.; Harrach, B.; Kaján, G.L.; Schlötterer, C.; Hess, M. The first whole genome sequence of a Fowl adenovirus B strain enables interspecies comparisons within the genus Aviadenovirus. Vet. Microbiol. 2013, 166, 250–256. [Google Scholar] [CrossRef]
- Hess, M. Commensal or pathogen—A challenge to fulfil Koch’s Postulates. Br. Poult. Sci. 2017, 58, 1–12. [Google Scholar] [CrossRef]
- Marek, A.; Günes, A.; Schulz, E.; Hess, M. Classification of fowl adenoviruses by use of phylogenetic analysis and high-resolution melting-curve analysis of the hexon L1 gene region. J. Virol. Methods 2010, 170, 147–154. [Google Scholar] [CrossRef]
- Kaján, G.L.; Kecskeméti, S.; Harrach, B.; Benkő, M. Molecular typing of fowl adenoviruses, isolated in Hungary recently, reveals high diversity. Vet. Microbiol. 2013, 167, 357–363. [Google Scholar] [CrossRef]
- Kaján, G.L.; Affranio, I.; Tóthné Bistyák, A.; Kecskeméti, S.; Benkő, M. An emerging new fowl adenovirus genotype. Heliyon 2019, 5, e01732. [Google Scholar] [CrossRef] [Green Version]
- Homonnay, Z.; Jakab, S.; Bali, K.; Kaszab, E.; Mató, T.; Kiss, I.; Palya, V.; Bányai, K. Genome sequencing of a novel variant of fowl adenovirus B reveals mosaicism in the pattern of homologous recombination events. Arch. Virol. 2021, 166, 1477–1480. [Google Scholar] [CrossRef] [PubMed]
- Meulemans, G.; Boschmans, M.; Berg, T.P.; Decaesstecker, M. Polymerase chain reaction combined with restriction enzyme analysis for detection and differentiation of fowl adenoviruses. Avian Pathol. 2001, 30, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Chevreux, B.; Wetter, T.; Suhai, S. Genome sequence assembly using trace signals and additional sequence information. In Proceedings of the German conference on bioinformatics, Heidelberg, Germany, 1999; Volume 99, pp. 45–56. [Google Scholar]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Lehwark, P.; Greiner, S. GB2sequin—A file converter preparing custom GenBank files for database submission. Genomics 2019, 111, 759–761. [Google Scholar] [CrossRef] [PubMed]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Schnurr, D. Neutralization. In Clinical Virology Manual; Specter, S.C., Hodinka, R.L., Young, S.A., Wiedbrauk, D.L., Eds.; ASM Press: Washington, DC, USA, 2009; ISBN 978-1-555-81462-5. [Google Scholar]
- Norrby, E. The structural and functional diversity of Adenovirus capsid components. J. Gen. Virol. 1969, 5, 221–236. [Google Scholar] [CrossRef]
- Song, Y.; Dimaio, F.; Wang, R.Y.R.; Kim, D.; Miles, C.; Brunette, T.; Thompson, J.; Baker, D. High-resolution comparative modeling with RosettaCM. Structure 2013, 21, 1735–1742. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger, L. Schrödinger Suite; Schrödinger, LLC: New York, NY, USA, 2021. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual MolecularDynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford-Miksza, L.; Schnurr, D.P. Analysis of 15 adenovirus hexon proteins reveals the location and structure of seven hypervariable regions containing serotype-specific residues. J. Virol. 1996, 70, 1836–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rux, J.J.; Kuser, P.R.; Burnett, R.M. Structural and phylogenetic analysis of adenovirus hexons by use of high-resolution X-ray crystallographic, molecular modeling, and sequence-based methods. J. Virol. 2003, 77, 9553–9566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradley, R.R.; Maxfield, L.F.; Lynch, D.M.; Iampietro, M.J.; Borducchi, E.N.; Barouch, D.H. Adenovirus serotype 5-specific neutralizing antibodies target multiple hexon hypervariable regions. J. Virol. 2012, 86, 1267–1272. [Google Scholar] [CrossRef] [Green Version]
- Bradley, R.R.; Lynch, D.M.; Iampietro, M.J.; Borducchi, E.N.; Barouch, D.H. Adenovirus Serotype 5 Neutralizing Antibodies Target both Hexon and Fiber following Vaccination and Natural Infection. J. Virol. 2012, 86, 625–629. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Duffy, M.R.; Deng, L.; Dakin, R.S.; Uil, T.; Custers, J.; Kelly, S.M.; McVey, J.H.; Nicklin, S.A.; Baker, A.H. Manipulating adenovirus hexon hypervariable loops dictates immune neutralisation and coagulation factor X-dependent cell interaction in vitro and in vivo. PLoS Pathog. 2015, 11, e1004673. [Google Scholar] [CrossRef] [Green Version]
- Nemerow, G.R.; Stewart, P.L.; Reddy, V.S. Structure of human adenovirus. Curr. Opin. Virol. 2012, 2, 115–121. [Google Scholar] [CrossRef] [Green Version]
- Gall, J.G.; Crystal, R.G.; Falck-Pedersen, E. Construction and characterization of hexon-chimeric adenoviruses: Specification of adenovirus serotype. J. Virol. 1998, 72, 10260–10264. [Google Scholar] [CrossRef] [Green Version]
- Schachner, A.; Gonzalez, G.; Endler, L.; Ito, K.; Hess, M. Fowl Adenovirus (FAdV) recombination with intertypic crossovers in genomes of FAdV-D and FAdV-E, displaying hybrid serological phenotypes. Viruses 2019, 11, 1094. [Google Scholar] [CrossRef] [Green Version]
- Gahéry-Ségard, H.; Farace, F.; Godfrin, D.; Gaston, J.; Lengagne, R.; Tursz, T.; Boulanger, P.; Guillet, J.-G. Immune Response to Recombinant Capsid Proteins of Adenovirus in Humans: Antifiber and Anti-Penton Base Antibodies Have a Synergistic Effect on Neutralizing Activity. J. Virol. 1998, 72, 2388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFerran, J.B.; McCracken, R.M.; Connor, T.J.; Evans, R.T. Isolation of viruses from clinical outbreaks of inclusion body hepatitis. Avian Pathol. 1976, 5, 315–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaján, G.L.; Sameti, S.; Benkő, M. Partial sequence of the DNA-dependent DNA polymerase gene of fowl adenoviruses: A reference panel for a general diagnostic PCR in poultry. Acta Vet. Hung. 2011, 59, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Kaján, G.L. Poultry adenoviruses. In Molecular Detection of Animal Viral Pathogens; Liu, D., Ed.; CRC Press: Boca Raton, FL, USA, 2016; pp. 735–746. ISBN 9781498700368. [Google Scholar]
- Gomis, S.; Goodhope, A.R.; Ojkic, A.D.; Willson, P. Inclusion body hepatitis as a primary disease in broilers in Saskatchewan, Canada. Avian Dis. 2006, 50, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Ojkić, D.; Martin, E.; Swinton, J.; Vaillancourt, J.P.; Boulianne, M.; Gomis, S. Genotyping of Canadian isolates of fowl adenoviruses. Avian Pathol. 2008, 37, 95–100. [Google Scholar] [CrossRef]
- Changjing, L.; Haiying, L.; Dongdong, W.; Jingjing, W.; Youming, W.; Shouchun, W.; Jida, L.; Ping, L.; Jianlin, W.; Shouzhen, X.; et al. Characterization of fowl adenoviruses isolated between 2007 and 2014 in China. Vet. Microbiol. 2016, 197, 62–67. [Google Scholar] [CrossRef]
- Niu, Y.; Sun, Q.; Zhang, G.; Sun, W.; Liu, X.; Xiao, Y.; Shang, Y.; Liu, S. Epidemiological investigation of outbreaks of fowl adenovirus infections in commercial chickens in China. Transbound. Emerg. Dis. 2018, 65, e121–e126. [Google Scholar] [CrossRef]
- De la Torre, D.; Nuñez, L.F.N.; Santander Parra, S.H.; Astolfi-Ferreira, C.S.; Piantino Ferreira, A.J. Molecular characterization of fowl adenovirus group I in commercial broiler chickens in Brazil. VirusDisease 2018, 29, 83–88. [Google Scholar] [CrossRef]
- Chen, L.; Yin, L.; Zhou, Q.; Peng, P.; Du, Y.; Liu, L.; Zhang, Y.; Xue, C.; Cao, Y. Epidemiological investigation of fowl adenovirus infections in poultry in China during 2015–2018. BMC Vet. Res. 2019, 15, 271. [Google Scholar] [CrossRef] [Green Version]
- Hess, M. Aviadenovirus infections. In Diseases of Poultry; Swayne, D.E., Ed.; Wiley-Blackwell: Oxford, UK, 2013; pp. 290–300. ISBN 978-0-470-95899-5. [Google Scholar]
- McCarthy, T.; Lebeck, M.G.; Capuano, A.W.; Schnurr, D.P.; Gray, G.C. Molecular typing of clinical adenovirus specimens by an algorithm which permits detection of adenovirus coinfections and intermediate adenovirus strains. J. Clin. Virol. 2009, 46, 80–84. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, H.; Shimizu, F.; Tsubahara, H. Avian adenovirus: Its properties and serological classification. Natl. Inst. Anim. Health Q. 1964, 4, 183–193. [Google Scholar]
- Meulemans, G.; Couvreur, B.; Decaesstecker, M.; Boschmans, M.; van den Berg, T.P. Phylogenetic analysis of fowl adenoviruses. Avian Pathol. 2004, 33, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Kaján, G.L.; Kecskeméti, S. First Hungarian isolation of an adenovirus type belonging to species Fowl adenovirus B. Magy. Állatorvosok Lapja 2011, 133, 347–352. [Google Scholar]
- Kleine, A.; Hafez, H.M.; Lüschow, D. Investigations on aviadenoviruses isolated from turkey flocks in Germany. Avian Pathol. 2016, 46, 181–187. [Google Scholar] [CrossRef]
- Niczyporuk, J.; Kozdrun, W.; Czekaj, H.; Piekarska, K.; Stys-Fijol, N. Characterisation of adenovirus strains represented species B and E isolated from broiler chicken flocks in eastern Poland. Heliyon 2021, 7, e06225. [Google Scholar] [CrossRef]
- Pauly, M.; Akoua-Koffi, C.; Buchwald, N.; Schubert, G.; Weiss, S.; Couacy-Hymann, E.; Anoh, A.E.; Mossoun, A.; Calvignac-Spencer, S.; Leendertz, S.A.; et al. Adenovirus in Rural Côte D‘Ivoire: High Diversity and Cross-Species Detection. Ecohealth 2015, 12, 441–452. [Google Scholar] [CrossRef] [Green Version]
- Lim, T.H.; Lee, H.J.; Lee, D.H.; Lee, Y.N.; Park, J.K.; Youn, H.N.; Kim, M.S.; Youn, H.S.; Lee, J.B.; Park, S.Y.; et al. Identification and virulence characterization of fowl adenoviruses in Korea. Avian Dis. 2011, 55, 554–560. [Google Scholar] [CrossRef]
- Dar, A.; Gomis, S.; Shirley, I.; Mutwiri, G.; Brownlie, R.; Potter, A.; Gerdts, V.; Tikoo, S.K. Pathotypic and molecular characterization of a fowl adenovirus associated with inclusion body hepatitis in Saskatchewan chickens. Avian Dis. 2012, 56, 73–81. [Google Scholar] [CrossRef]
- Cui, J.; Xu, Y.; Zhou, Z.; Xu, Q.; Wang, J.; Xiao, Y.; Li, Z.; Bi, D. Pathogenicity and Molecular Typing of Fowl Adenovirus-Associated With Hepatitis/Hydropericardium Syndrome in Central China (2015–2018). Front. Vet. Sci. 2020, 7, 190. [Google Scholar] [CrossRef]
- Chitradevi, S.; Sukumar, K.; Suresh, P.; Balasubramaniam, G.A.; Kannan, D. Molecular typing and pathogenicity assessment of fowl adenovirus associated with inclusion body hepatitis in chicken from India. Trop. Anim. Health Prod. 2021, 53, 412. [Google Scholar] [CrossRef]
- Steer, P.A.; O’Rourke, D.; Ghorashi, S.A.; Noormohammadi, A.H. Application of high-resolution melting curve analysis for typing of fowl adenoviruses in field cases of inclusion body hepatitis. Aust. Vet J. 2011, 89, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Niczyporuk, J.S. Phylogenetic and geographic analysis of fowl adenovirus field strains isolated from poultry in Poland. Arch. Virol. 2016, 161, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Niczyporuk, J.S.; Czekaj, H. A comparative pathogenicity analysis of two adenovirus strains, 1/A and 8a/E, isolated from poultry in Poland. Arch. Virol. 2018, 163, 3005–3013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redondo, H.; Fragoso, J.S.; Tahala, M.A.; Bensassi, Y.; Gil, I.; Elbachir, E.; Rodríguez, M.J.; Moreno, J.C.A. Characterization of strain of fowl adenoviruses circulating in Morocco. Poult. Sci. 2018, 97, 4057–4062. [Google Scholar] [CrossRef]
- Oliver-Ferrando, S.; Dolz, R.; Calderón, C.; Valle, R.; Rivas, R.; Pérez, M.; Biarnés, M.; Blanco, A.; Bertran, K.; Ramis, A.; et al. Epidemiological and pathological investigation of fowl aviadenovirus serotypes 8b and 11 isolated from chickens with inclusion body hepatitis in Spain (2011–2013). Avian Pathol. 2017, 46, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Morshed, R.; Hosseini, H.; Langeroudi, A.G.; Fard, M.H.B.; Charkhkar, S. Fowl Adenoviruses D and e Cause Inclusion Body Hepatitis Outbreaks in Broiler and Broiler Breeder Pullet Flocks. Avian Dis. 2017, 61, 205–210. [Google Scholar] [CrossRef]
- McFerran, J.B.; Connor, T.J.; McCracken, R.M. Isolation of adenoviruses and reoviruses from avian species other than domestic fowl. Avian Dis. 1976, 20, 519–524. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).