
Chikungunya Virus’ High Genomic Plasticity Enables Rapid Adaptation to Restrictive A549 Cells

 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Passaging of CHIKV
2.3. Luciferase Assay
2.4. Deep Sequence Analysis
2.5. Molecular Cloning
2.6. Trans-Replicase Assay (TRA)
2.7. Plaque Assay
2.8. Binding Assay
3. Results
3.1. CHIKV Replication Is Restricted in A549 Cells
3.2. Adaptation of CHIKV on A549 Cells
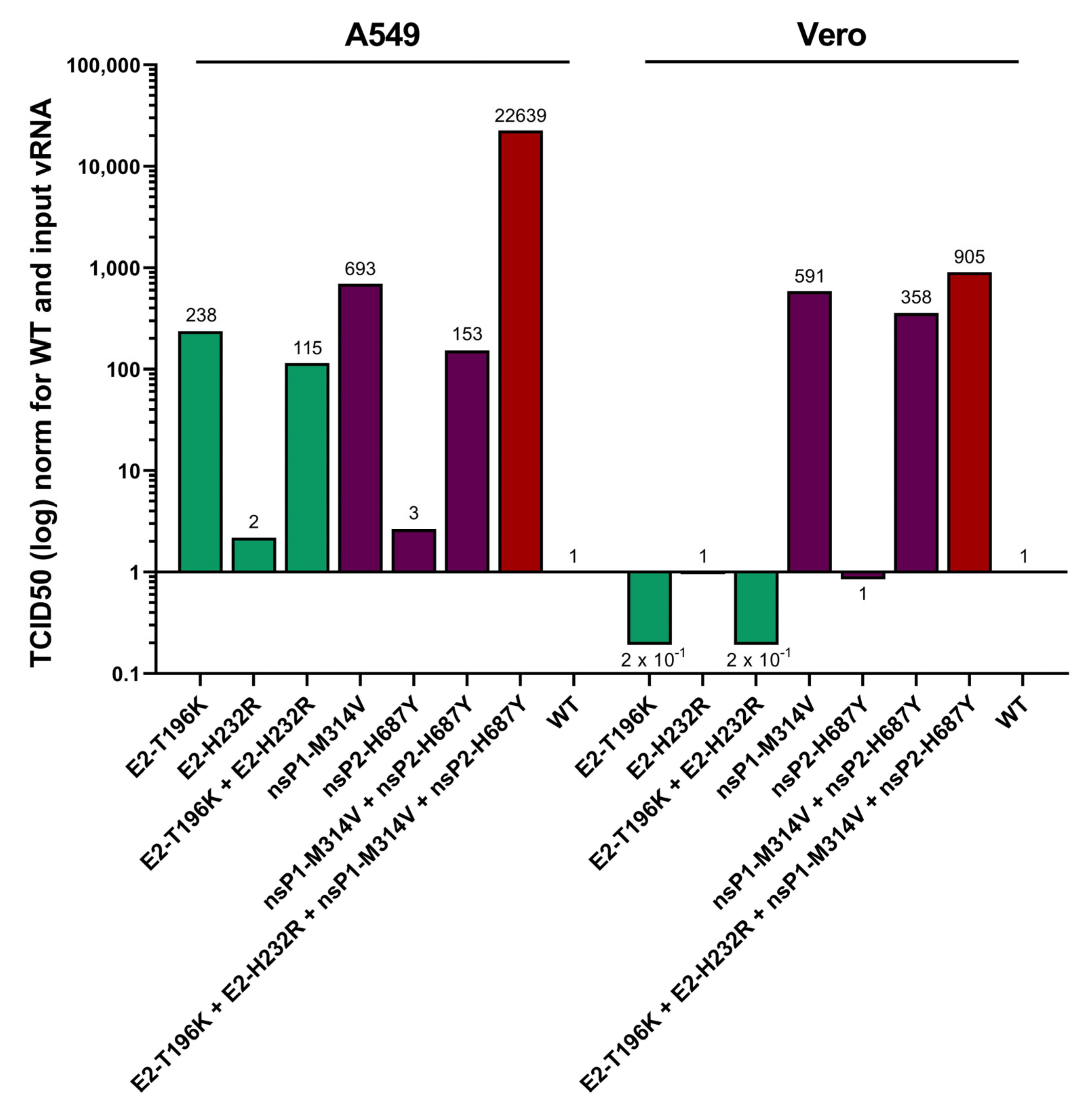
3.3. Contribution of the Identified Mutations to the Viral Phenotype
3.4. Mutations in nsP’s Have a Role Outside the Replicase Complex
3.5. Adaptive Mutations in the Envelope E2 Protein Enhance Attachment to Both A549 and Vero Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Solignat, M.; Gay, B.; Higgs, S.; Briant, L.; Devaux, C. Replication cycle of chikungunya: A re-emerging arbovirus. Virology 2009, 393, 183–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lumsden, W.H.R. An epidemic of virus disease in Southern Province, Tanganyika territory, in 1952–1953 II. General description and epidemiology. Trans. R. Soc. Trop. Med. Hyg. 1955, 49, 33–57. [Google Scholar] [CrossRef]
- Mascarenhas, M.; Garasia, S.; Berthiaume, P.; Corrin, T.; Greig, J.; Ng, V.; Young, I.; Waddell, L. A scoping review of published literature on chikungunya virus. PLoS ONE 2018, 13, e0207554. [Google Scholar] [CrossRef] [PubMed]
- Thiboutot, M.M.; Kannan, S.; Kawalekar, O.U.; Shedlock, D.J.; Khan, A.S.; Sarangan, G.; Srikanth, P.; Weiner, D.B.; Muthumani, K. Chikungunya: A potentially emerging epidemic? PLoS Negl. Trop. Dis. 2010, 4, e623. [Google Scholar] [CrossRef]
- Burt, F.J.; Chen, W.; Miner, J.J.; Lenschow, D.J.; Merits, A.; Schnettler, E.; Kohl, A.; Rudd, P.A.; Taylor, A.; Herrero, L.J.; et al. Chikungunya virus: An update on the biology and pathogenesis of this emerging pathogen. Lancet Infect. Dis. 2017, 17, e107–e117. [Google Scholar] [CrossRef]
- Tsetsarkin, K.A.; Chen, R.; Weaver, S.C. Interspecies transmission and chikungunya virus emergence. Curr. Opin. Virol. 2016, 16, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Weaver, S.C.; Forrester, N.L. Chikungunya: Evolutionary history and recent epidemic spread. Antivir. Res. 2015, 120, 32–39. [Google Scholar] [CrossRef]
- Tsetsarkin, K.A.; Vanlandingham, D.L.; McGee, C.E.; Higgs, S. A Single Mutation in Chikungunya Virus Affects Vector Specificity and Epidemic Potential. PLOS Pathog. 2007, 3, e201. [Google Scholar] [CrossRef]
- Tsetsarkin, K.A.; Chen, R.; Yun, R.; Rossi, S.L.; Plante, K.S.; Guerbois, M.; Forrester, N.; Perng, G.C.; Sreekumar, E.; Leal, G.; et al. Multi-peaked adaptive landscape for chikungunya virus evolution predicts continued fitness optimization in Aedes albopictus mosquitoes. Nat. Commun. 2014, 5, 4084. [Google Scholar] [CrossRef] [Green Version]
- Tsetsarkin, K.A.; McGee, C.E.; Volk, S.M.; Vanlandingham, D.L.; Weaver, S.C.; Higgs, S. Epistatic roles of E2 glycoprotein mutations in adaption of chikungunya virus to Aedes albopictus and Ae. aegypti mosquitoes. PLoS ONE 2009, 4, e6835. [Google Scholar] [CrossRef] [Green Version]
- Tsetsarkin, K.A.; Weaver, S.C. Sequential adaptive mutations enhance efficient vector switching by Chikungunya virus and its epidemic emergence. PLoS Pathog. 2011, 7, e1002412. [Google Scholar] [CrossRef] [Green Version]
- Tsetsarkin, K.A.; Chen, R.; Leal, G.; Forrester, N.; Higgs, S.; Huang, J.; Weaver, S.C. Chikungunya virus emergence is constrained in Asia by lineage-specific adaptive landscapes. Proc. Natl. Acad. Sci. USA 2011, 108, 7872–7877. [Google Scholar] [CrossRef] [Green Version]
- Matusali, G.; Colavita, F.; Bordi, L.; Lalle, E.; Ippolito, G.; Capobianchi, M.R.; Castilletti, C. Tropism of the Chikungunya Virus. Viruses 2019, 11, 175. [Google Scholar] [CrossRef] [Green Version]
- Couderc, T.; Lecuit, M. Chikungunya virus pathogenesis: From bedside to bench. Antivir. Res. 2015, 121, 120–131. [Google Scholar] [CrossRef]
- Roberts, G.C.; Zothner, C.; Remenyi, R.; Merits, A.; Stonehouse, N.J.; Harris, M. Evaluation of a range of mammalian and mosquito cell lines for use in Chikungunya virus research. Sci. Rep. 2017, 7, 14641. [Google Scholar] [CrossRef] [Green Version]
- Weber, C.; König, R.; Niedrig, M.; Emmerich, P.; Schnierle, B.S. A neutralization assay for chikungunya virus infections in a multiplex format. J. Virol. Methods 2014, 201, 7–12. [Google Scholar] [CrossRef]
- Salvador, B.; Zhou, Y.; Michault, A.; Muench, M.O.; Simmons, G. Characterization of Chikungunya pseudotyped viruses: Identification of refractory cell lines and demonstration of cellular tropism differences mediated by mutations in E1 glycoprotein. Virology 2009, 393, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Sourisseau, M.; Schilte, C.; Casartelli, N.; Trouillet, C.; Guivel-Benhassine, F.; Rudnicka, D.; Sol-Foulon, N.; Le Roux, K.; Prevost, M.-C.; Fsihi, H.; et al. Characterization of reemerging chikungunya virus. PLoS Pathog. 2007, 3, e89. [Google Scholar] [CrossRef]
- De Caluwé, L.; Coppens, S.; Vereecken, K.; Daled, S.; Dhaenens, M.; Van Ostade, X.; Deforce, D.; Ariën, K.K.; Bartholomeeusen, K. The CD147 Protein Complex Is Involved in Entry of Chikungunya Virus and Related Alphaviruses in Human Cells. Front. Microbiol. 2021, 12, 615165. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishnan, M.A. Determination of 50% endpoint titer using a simple formula. World J. Virol. 2016, 5, 85–86. [Google Scholar] [CrossRef] [PubMed]
- Utt, A.; Rausalu, K.; Jakobson, M.; Männik, A.; Alphey, L.; Fragkoudis, R.; Merits, A.; Dermody, T.S. Design and Use of Chikungunya Virus Replication Templates Utilizing Mammalian and Mosquito RNA Polymerase I-Mediated Transcription. J. Virol. 2019, 93, e00794-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartholomeeusen, K.; Utt, A.; Coppens, S.; Rausalu, K.; Vereecken, K.; Ariën, K.K.; Merits, A. A Chikungunya Virus trans-Replicase System Reveals the Importance of Delayed Nonstructural Polyprotein Processing for Efficient Replication Complex Formation in Mosquito Cells. J. Virol. 2018, 92, e00152-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utt, A.; Quirin, T.; Saul, S.; Hellström, K.; Ahola, T.; Merits, A. Versatile Trans-Replication Systems for Chikungunya Virus Allow Functional Analysis and Tagging of Every Replicase Protein. PLoS ONE 2016, 11, e0151616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashbrook, A.W.; Burrack, K.S.; Silva, L.A.; Montgomery, S.A.; Heise, M.T.; Morrison, T.E.; Dermody, T.S. Residue 82 of the Chikungunya virus E2 attachment protein modulates viral dissemination and arthritis in mice. J. Virol. 2014, 88, 12180–12192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, L.A.; Khomandiak, S.; Ashbrook, A.W.; Weller, R.; Heise, M.T.; Morrison, T.E.; Dermody, T.S. A single-amino-acid polymorphism in Chikungunya virus E2 glycoprotein influences glycosaminoglycan utilization. J. Virol. 2014, 88, 2385–2397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henrik Gad, H.; Paulous, S.; Belarbi, E.; Diancourt, L.; Drosten, C.; Kummerer, B.M.; Plate, A.E.; Caro, V.; Despres, P. The E2-E166K substitution restores Chikungunya virus growth in OAS3 expressing cells by acting on viral entry. Virology 2012, 434, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Weber, C.; Berberich, E.; von Rhein, C.; Henß, L.; Hildt, E.; Schnierle, B.S. Identification of Functional Determinants in the Chikungunya Virus E2 Protein. PLoS Negl. Trop. Dis. 2017, 11, e0005318. [Google Scholar] [CrossRef]
- Kamhi, E.; Joo, E.J.; Dordick, J.S.; Linhardt, R.J. Glycosaminoglycans in infectious disease. Biol. Rev. 2013, 88, 928–943. [Google Scholar] [CrossRef]
- Voss, J.E.; Vaney, M.C.; Duquerroy, S.; Vonrhein, C.; Girard-Blanc, C.; Crublet, E.; Thompson, A.; Bricogne, G.; Rey, F.A. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 2010, 468, 709–712. [Google Scholar] [CrossRef]
- Basore, K.; Kim, A.S.; Nelson, C.A.; Zhang, R.; Smith, B.K.; Uranga, C.; Vang, L.; Cheng, M.; Gross, M.L.; Smith, J.; et al. Cryo-EM Structure of Chikungunya Virus in Complex with the Mxra8 Receptor. Cell 2019, 177, 1725–1737.e1716. [Google Scholar] [CrossRef]
- Song, H.; Zhao, Z.; Chai, Y.; Jin, X.; Li, C.; Yuan, F.; Liu, S.; Gao, Z.; Wang, H.; Song, J.; et al. Molecular Basis of Arthritogenic Alphavirus Receptor MXRA8 Binding to Chikungunya Virus Envelope Protein. Cell 2019, 177, 1714–1724.e1712. [Google Scholar] [CrossRef]
- Zhang, R.; Kim, A.S.; Fox, J.M.; Nair, S.; Basore, K.; Klimstra, W.B.; Rimkunas, R.; Fong, R.H.; Lin, H.; Poddar, S.; et al. Mxra8 is a receptor for multiple arthritogenic alphaviruses. Nature 2018, 557, 570–574. [Google Scholar] [CrossRef]
- Zhang, R.; Earnest, J.T.; Kim, A.S.; Winkler, E.S.; Desai, P.; Adams, L.J.; Hu, G.; Bullock, C.; Gold, B.; Cherry, S.; et al. Expression of the Mxra8 Receptor Promotes Alphavirus Infection and Pathogenesis in Mice and Drosophila. Cell Rep. 2019, 28, 2647–2658.e2645. [Google Scholar] [CrossRef]
- Kim, A.S.; Zimmerman, O.; Fox, J.M.; Nelson, C.A.; Basore, K.; Zhang, R.; Durnell, L.; Desai, C.; Bullock, C.; Deem, S.L.; et al. An Evolutionary Insertion in the Mxra8 Receptor-Binding Site Confers Resistance to Alphavirus Infection and Pathogenesis. Cell Host Microbe 2020, 27, 428–440.e429. [Google Scholar] [CrossRef]
- Sun, S.; Xiang, Y.; Akahata, W.; Holdaway, H.; Pal, P.; Zhang, X.; Diamond, M.S.; Nabel, G.J.; Rossmann, M.G. Structural analyses at pseudo atomic resolution of Chikungunya virus and antibodies show mechanisms of neutralization. Elife 2013, 2, e00435. [Google Scholar] [CrossRef]
- Zhou, Q.F.; Fox, J.M.; Earnest, J.T.; Ng, T.-S.; Kim, A.S.; Fibriansah, G.; Kostyuchenko, V.A.; Shi, J.; Shu, B.; Diamond, M.S.; et al. Structural basis of Chikungunya virus inhibition by monoclonal antibodies. Proc. Natl. Acad. Sci. USA 2020, 117, 27637–27645. [Google Scholar] [CrossRef]
- Fox, J.M.; Long, F.; Edeling, M.A.; Lin, H.; van Duijl-Richter, M.K.S.; Fong, R.H.; Kahle, K.M.; Smit, J.M.; Jin, J.; Simmons, G.; et al. Broadly Neutralizing Alphavirus Antibodies Bind an Epitope on E2 and Inhibit Entry and Egress. Cell 2015, 163, 1095–1107. [Google Scholar] [CrossRef] [Green Version]
- Ahola, T.; Merits, A. Functions of Chikungunya Virus Nonstructural Proteins. In Chikungunya Virus: Advances in Biology, Pathogenesis, and Treatment; Okeoma, C.M., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 75–98. [Google Scholar] [CrossRef]
- Jones, P.H.; Maric, M.; Madison, M.N.; Maury, W.; Roller, R.J.; Okeoma, C.M. BST-2/tetherin-mediated restriction of chikungunya (CHIKV) VLP budding is counteracted by CHIKV non-structural protein 1 (nsP1). Virology 2013, 438, 37–49. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.; Bragagnolo, G.; Arranz, R.; Reguera, J. Capping pores of alphavirus nsP1 gate membranous viral replication factories. Nature 2021, 589, 615–619. [Google Scholar] [CrossRef]
- Kumar, S.; Kumar, A.; Mamidi, P.; Tiwari, A.; Kumar, S.; Mayavannan, A.; Mudulli, S.; Singh, A.K.; Subudhi, B.B.; Chattopadhyay, S. Chikungunya virus nsP1 interacts directly with nsP2 and modulates its ATPase activity. Sci. Rep. 2018, 8, 1045. [Google Scholar] [CrossRef] [Green Version]
- Volk, S.M.; Chen, R.; Tsetsarkin, K.A.; Adams, A.P.; Garcia, T.I.; Sall, A.A.; Nasar, F.; Schuh, A.J.; Holmes, E.C.; Higgs, S.; et al. Genome-scale phylogenetic analyses of chikungunya virus reveal independent emergences of recent epidemics and various evolutionary rates. J. Virol. 2010, 84, 6497–6504. [Google Scholar] [CrossRef] [Green Version]
- Frolov, I.; Garmashova, N.; Atasheva, S.; Frolova, E.I. Random insertion mutagenesis of sindbis virus nonstructural protein 2 and selection of variants incapable of downregulating cellular transcription. J. Virol. 2009, 83, 9031–9044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, J.; Rajasekharan, S.; Gulati, S.; Dudha, N.; Gupta, A.; Chaudhary, V.K.; Gupta, S. Network mapping among the functional domains of Chikungunya virus nonstructural proteins. Proteins Struct. Funct. Bioinform. 2014, 82, 2403–2411. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 5′ Primer F | 3′ Primer R | Fragment |
|---|---|---|
| ATGGCTGCGTGAGACACACGTAGC | TTGCTTCATCCAGCTTAGGTGGG | 19—5816 |
| AGCGACTGGTCCACGTGCT | GAAATATTAAAAACAAAATAACATCTCCTACGTCCCTGT | 5642—11829 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Caluwé, L.; Heyndrickx, L.; Coppens, S.; Vereecken, K.; Quiñones-Mateu, M.E.; Merits, A.; Ariën, K.K.; Bartholomeeusen, K. Chikungunya Virus’ High Genomic Plasticity Enables Rapid Adaptation to Restrictive A549 Cells. Viruses 2022, 14, 282. https://doi.org/10.3390/v14020282
De Caluwé L, Heyndrickx L, Coppens S, Vereecken K, Quiñones-Mateu ME, Merits A, Ariën KK, Bartholomeeusen K. Chikungunya Virus’ High Genomic Plasticity Enables Rapid Adaptation to Restrictive A549 Cells. Viruses. 2022; 14(2):282. https://doi.org/10.3390/v14020282
Chicago/Turabian StyleDe Caluwé, Lien, Leo Heyndrickx, Sandra Coppens, Katleen Vereecken, Miguel E. Quiñones-Mateu, Andres Merits, Kevin K. Ariën, and Koen Bartholomeeusen. 2022. "Chikungunya Virus’ High Genomic Plasticity Enables Rapid Adaptation to Restrictive A549 Cells" Viruses 14, no. 2: 282. https://doi.org/10.3390/v14020282
APA StyleDe Caluwé, L., Heyndrickx, L., Coppens, S., Vereecken, K., Quiñones-Mateu, M. E., Merits, A., Ariën, K. K., & Bartholomeeusen, K. (2022). Chikungunya Virus’ High Genomic Plasticity Enables Rapid Adaptation to Restrictive A549 Cells. Viruses, 14(2), 282. https://doi.org/10.3390/v14020282