
Computational Analysis of Mutations in the Receptor-Binding Domain of SARS-CoV-2 Spike and Their Effects on Antibody Binding

 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Analysis of Atomic Structures and Clustering
2.2. Estimation of Binding Energies
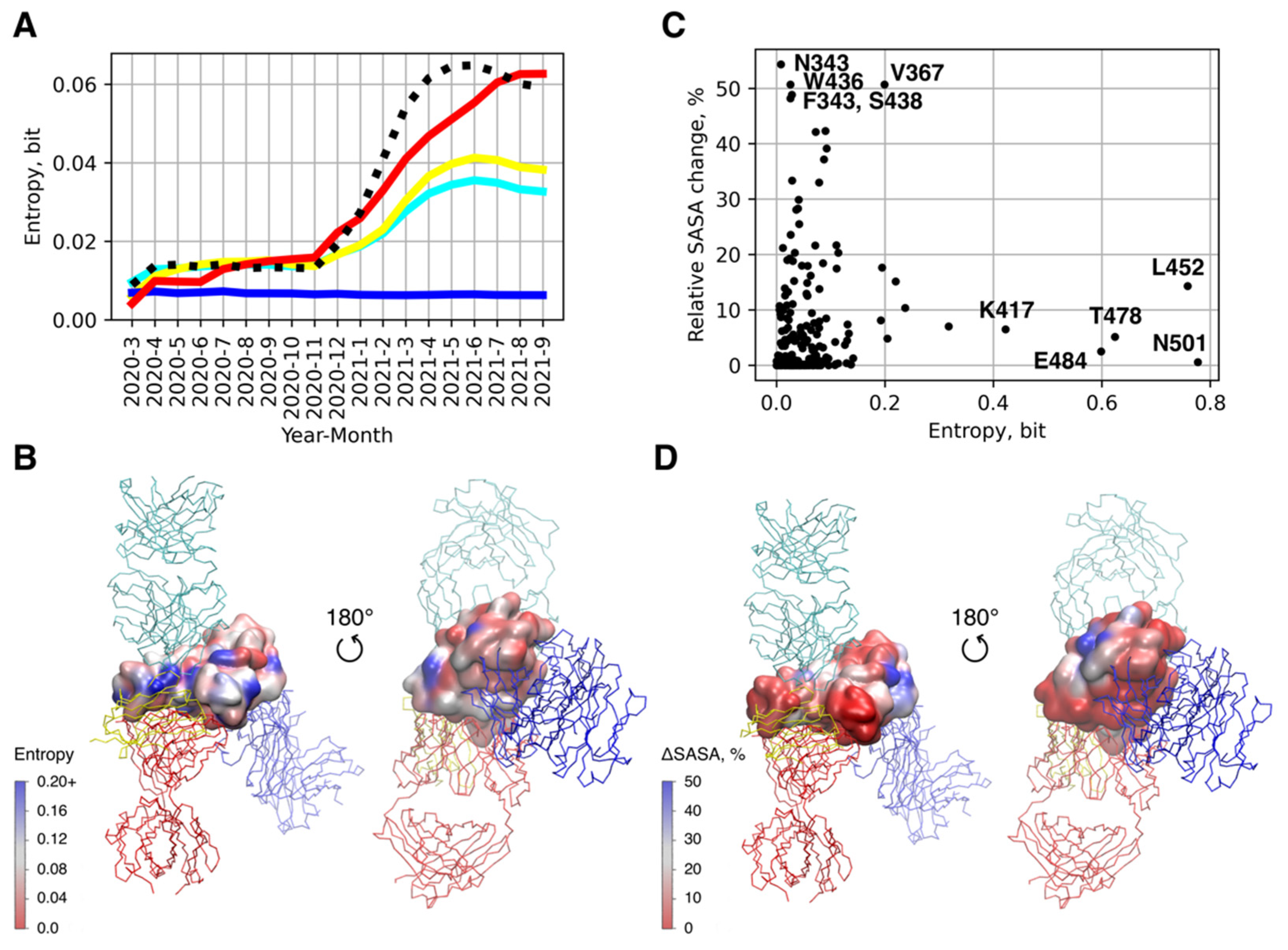
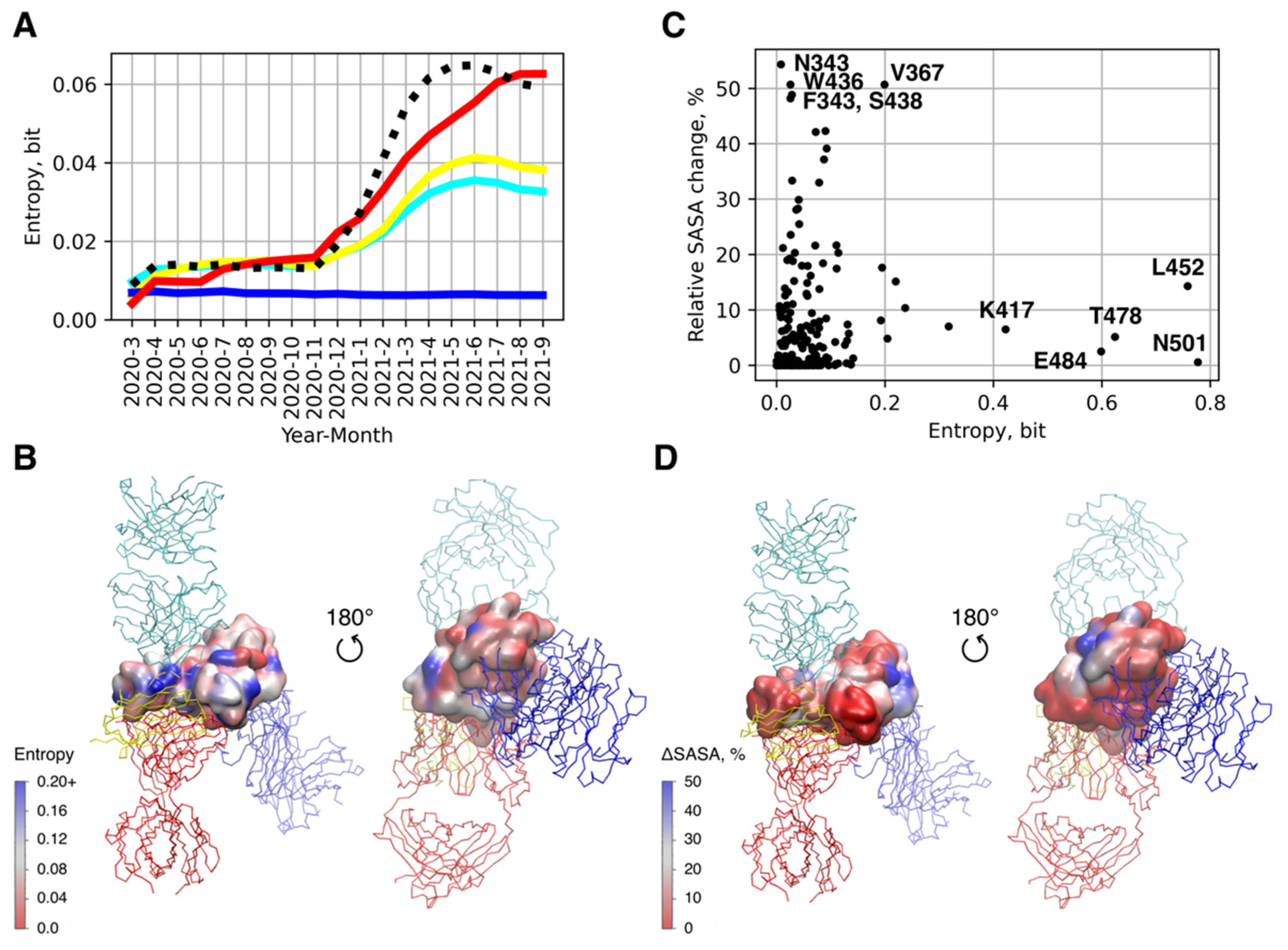
2.3. Sequence Analysis
2.4. Analysis of Epitope Accessibility in the Glycolisalted S Protein
3. Results and Discussion
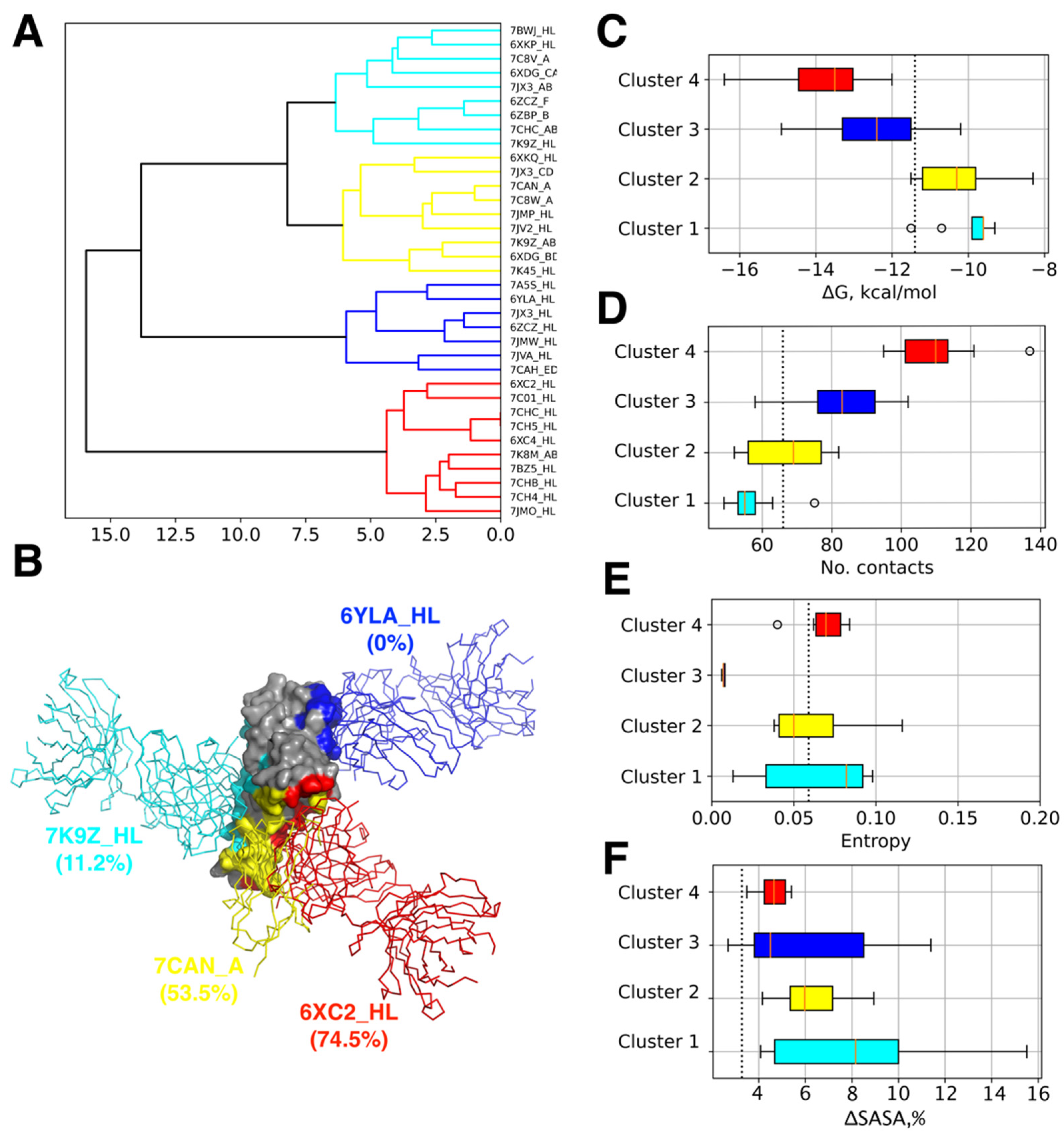
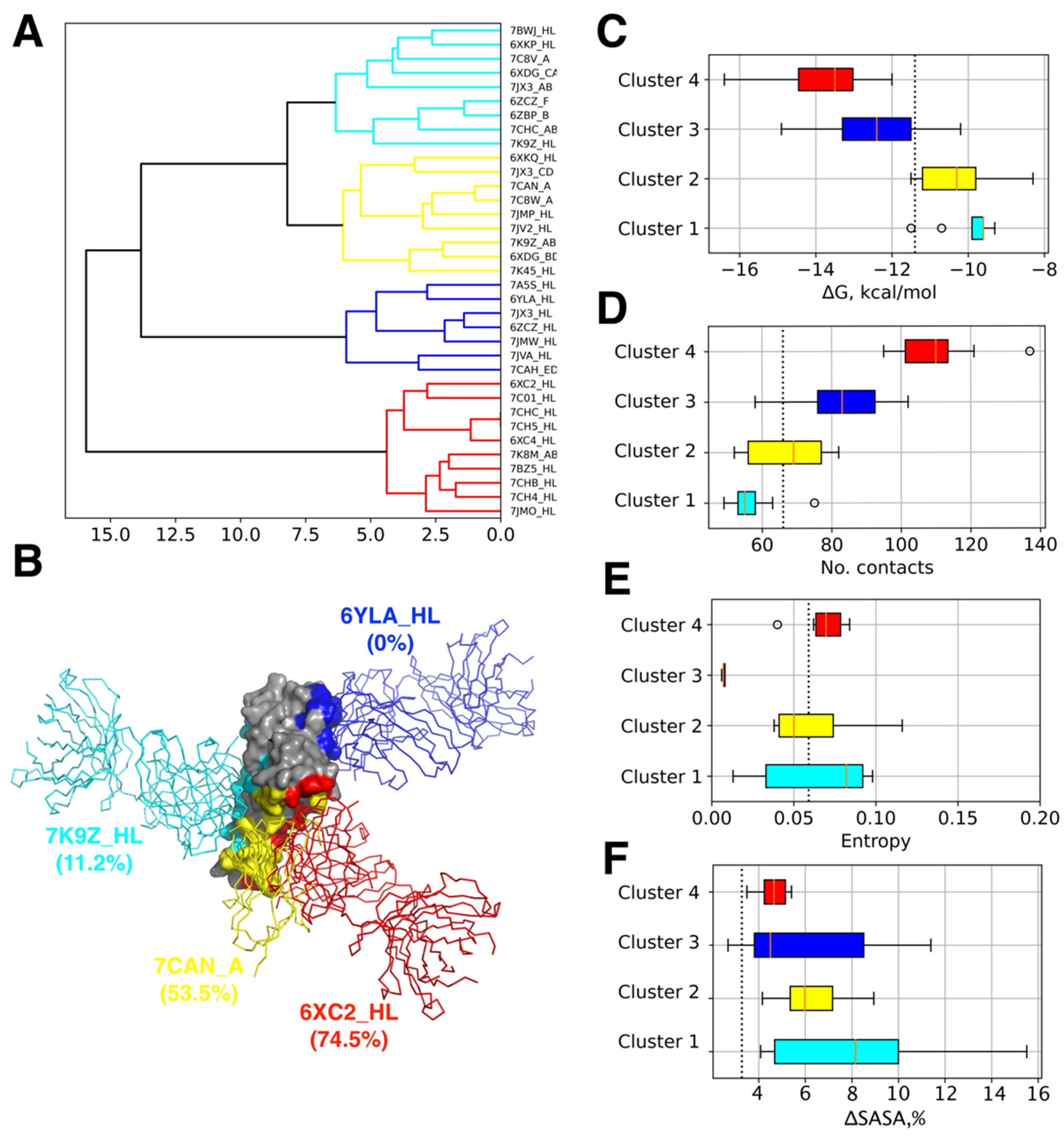
3.1. Analysis of Epitopes Reveals Distinct Clusters of Ab Binding Poses with Unique Features
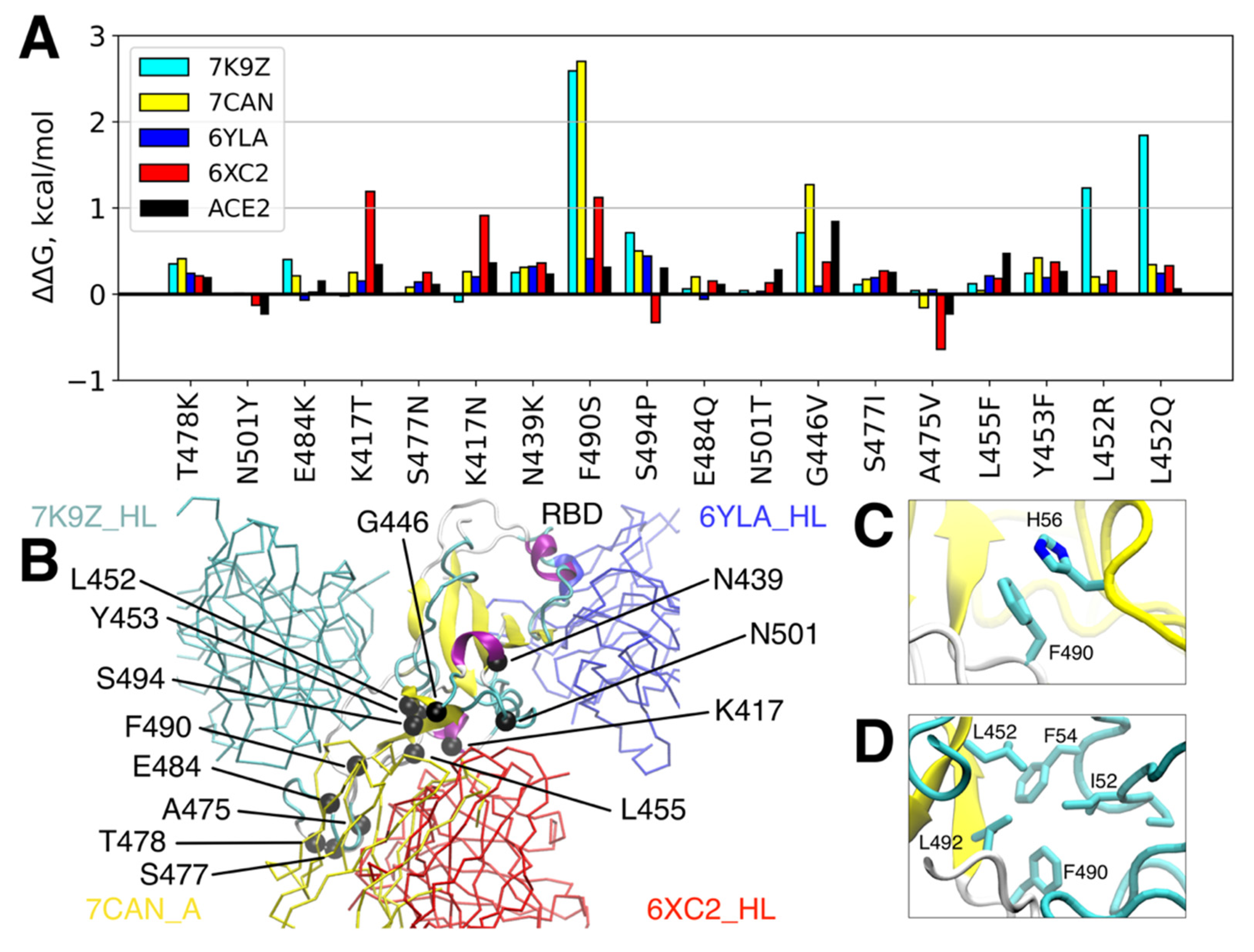
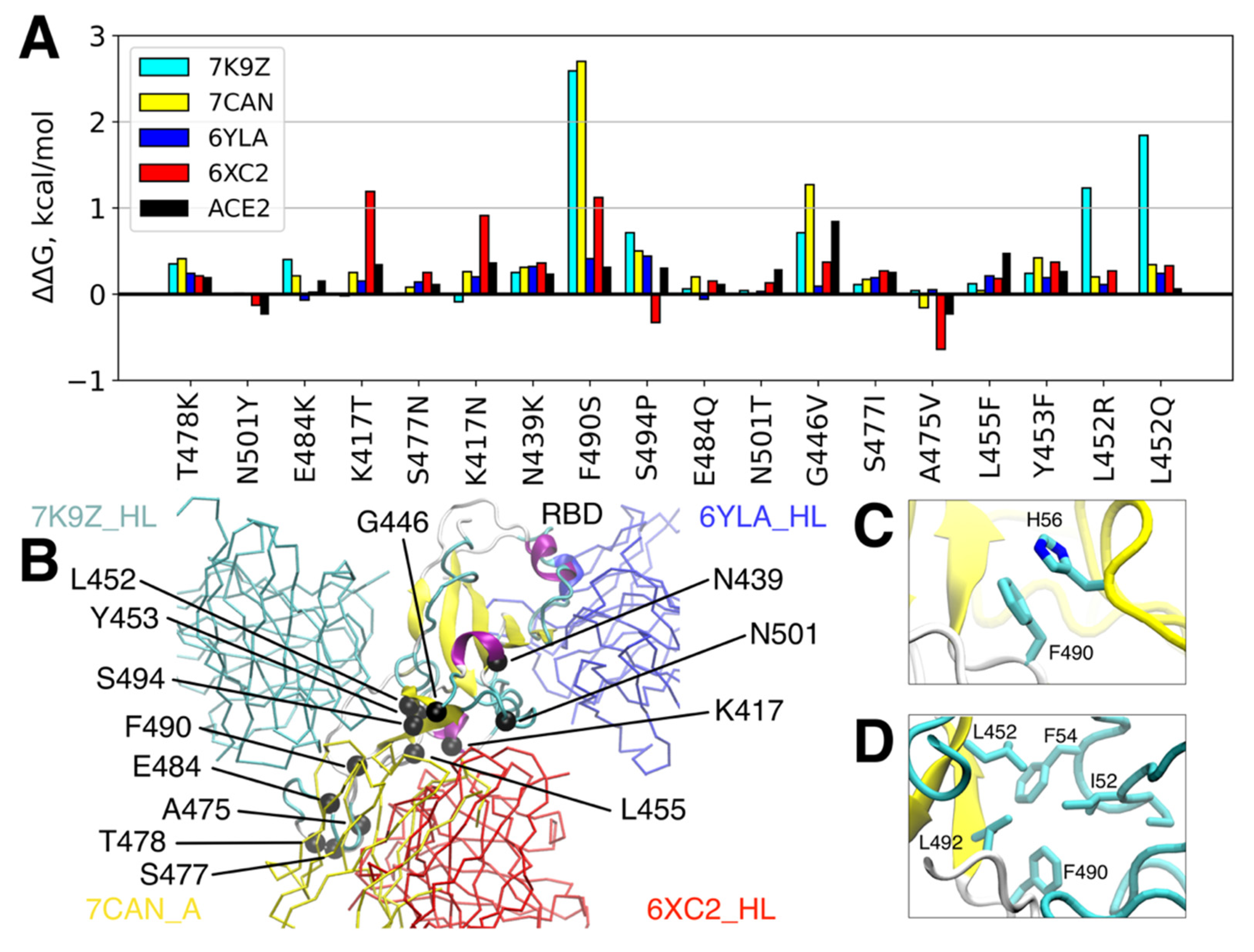
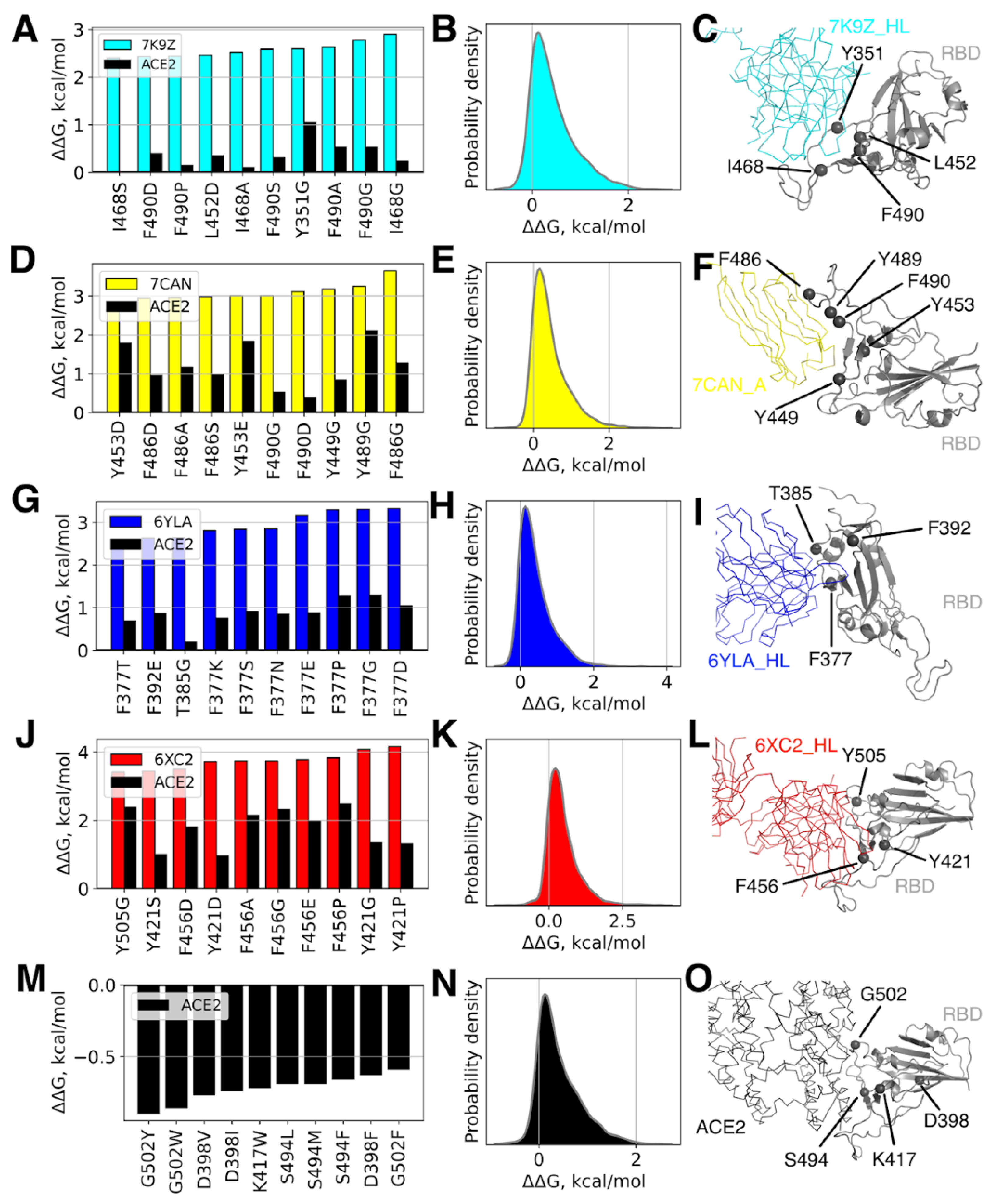
3.2. Single RBD Mutations Have Limited Effect on its Binding Affinity to Abs and ACE2
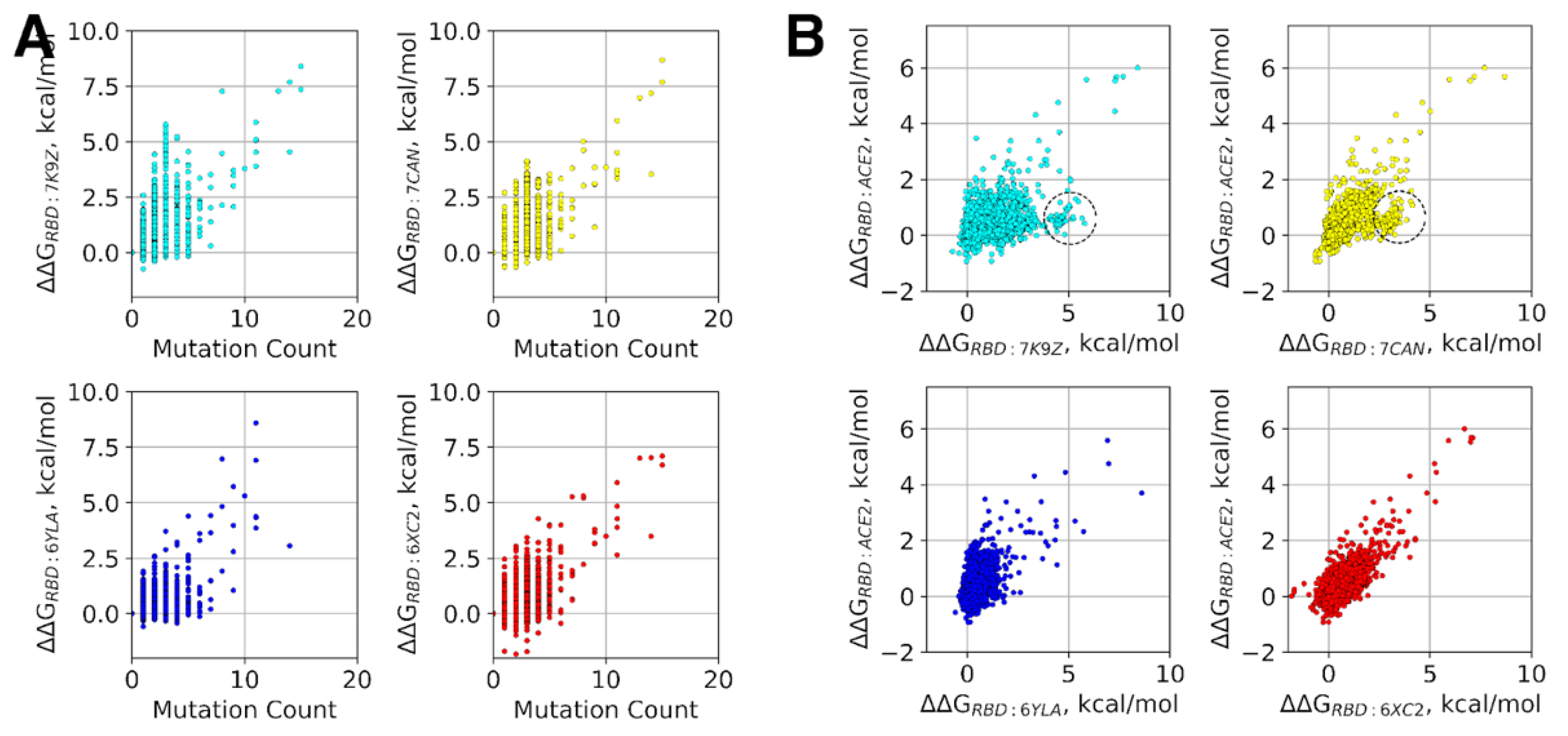
3.3. Antigenic Escape Assessment for Existing SARS-CoV2 Variants with Multiple Mutations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Krammer, F. SARS-CoV-2 Vaccines in Development. Nature 2020, 586, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Xiaojie, S.; Yu, L.; Lei, Y.; Guang, Y.; Min, Q. Neutralizing Antibodies Targeting SARS-CoV-2 Spike Protein. Stem Cell Res. 2020, 50, 102125. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-Converting Enzyme 2 Is a Functional Receptor for the SARS Coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural Basis of Receptor Recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 Entry into Cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Chi, X.; Yan, R.; Zhang, J.; Zhang, G.; Zhang, Y.; Hao, M.; Zhang, Z.; Fan, P.; Dong, Y.; Yang, Y.; et al. A Neutralizing Human Antibody Binds to the N-Terminal Domain of the Spike Protein of SARS-CoV-2. Science 2020, 369, 650–655. [Google Scholar] [CrossRef]
- Tong, P.; Gautam, A.; Windsor, I.W.; Travers, M.; Chen, Y.; Garcia, N.; Whiteman, N.B.; McKay, L.G.A.; Storm, N.; Malsick, L.E.; et al. Memory B Cell Repertoire for Recognition of Evolving SARS-CoV-2 Spike. Cell 2021, 184, 4969–4980.e15. [Google Scholar] [CrossRef]
- Gaebler, C.; Wang, Z.; Lorenzi, J.C.C.; Muecksch, F.; Finkin, S.; Tokuyama, M.; Cho, A.; Jankovic, M.; Schaefer-Babajew, D.; Oliveira, T.Y.; et al. Evolution of Antibody Immunity to SARS-CoV-2. Nature 2021, 591, 639–644. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; COVID-19 Genomics UK (COG-UK) Consortium; et al. SARS-CoV-2 Variants, Spike Mutations and Immune Escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Alam, I.; Radovanovic, A.; Incitti, R.; Kamau, A.A.; Alarawi, M.; Azhar, E.I.; Gojobori, T. CovMT: An Interactive SARS-CoV-2 Mutation Tracker, with a Focus on Critical Variants. Lancet Infect. Dis. 2021, 21, 602. [Google Scholar] [CrossRef]
- Wang, P.; Nair, M.S.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; et al. Antibody Resistance of SARS-CoV-2 Variants B.1.351 and B.1.1.7. Nature 2021, 593, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Dawood, A.A. Mutated COVID. New Microbes New Infect. 2020, 35, 100673. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and Evolution of Pathogenic Coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisblum, Y.; Schmidt, F.; Zhang, F.; DaSilva, J.; Poston, D.; Lorenzi, J.C.; Muecksch, F.; Rutkowska, M.; Hoffmann, H.-H.; Michailidis, E.; et al. Escape from Neutralizing Antibodies by SARS-CoV-2 Spike Protein Variants. eLife 2020, 9, e61312. [Google Scholar] [CrossRef] [PubMed]
- Satyam, R.; Yousef, M.; Qazi, S.; Bhat, A.M.; Raza, K. COVIDium: A COVID-19 Resource Compendium. Database 2021, 2021, baab057. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, M.A.; Liu, Y.; Feng, C.; Hofestädt, R.; Chen, M. OverCOVID: An Integrative Web Portal for SARS-CoV-2 Bioinformatics Resources. J. Integr. Bioinform. 2021, 18, 9–17. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global Initiative on Sharing All Influenza Data—From Vision to Reality. Euro Surveill. 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- Ahsan, M.A.; Liu, Y.; Feng, C.; Zhou, Y.; Ma, G.; Bai, Y.; Chen, M. Bioinformatics Resources Facilitate Understanding and Harnessing Clinical Research of SARS-CoV-2. Brief. Bioinform. 2021, 22, 714–725. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Addetia, A.; Hannon, W.W.; Choudhary, M.C.; Dingens, A.S.; Li, J.Z.; Bloom, J.D. Prospective Mapping of Viral Mutations That Escape Antibodies Used to Treat COVID-19. Science 2021, 371, 850–854. [Google Scholar] [CrossRef]
- Thomson, E.C.; Rosen, L.E.; Shepherd, J.G.; Spreafico, R.; da Silva Filipe, A.; Wojcechowskyj, J.A.; Davis, C.; Piccoli, L.; Pascall, D.J.; Dillen, J.; et al. Circulating SARS-CoV-2 Spike N439K Variants Maintain Fitness While Evading Antibody-Mediated Immunity. Cell 2021, 184, 1171.e20–1187.e20. [Google Scholar] [CrossRef]
- Li, Q.; Nie, J.; Wu, J.; Zhang, L.; Ding, R.; Wang, H.; Zhang, Y.; Li, T.; Liu, S.; Zhang, M.; et al. No Higher Infectivity but Immune Escape of SARS-CoV-2 501Y.V2 Variants. Cell 2021, 184, 2362–2371. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM Structure of the 2019-NCoV Spike in the Prefusion Conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281.e6–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural Basis for the Recognition of SARS-CoV-2 by Full-Length Human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, J.; Baum, A.; Pascal, K.E.; Russo, V.; Giordano, S.; Wloga, E.; Fulton, B.O.; Yan, Y.; Koon, K.; Patel, K.; et al. Studies in Humanized Mice and Convalescent Humans Yield a SARS-CoV-2 Antibody Cocktail. Science 2020, 369, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Kreye, J.; Reincke, S.M.; Kornau, H.-C.; Sánchez-Sendin, E.; Corman, V.M.; Liu, H.; Yuan, M.; Wu, N.C.; Zhu, X.; Lee, C.-C.D.; et al. A Therapeutic Non-Self-Reactive SARS-CoV-2 Antibody Protects from Lung Pathology in a COVID-19 Hamster Model. Cell 2020, 183, 1058.e19–1069.e19. [Google Scholar] [CrossRef] [PubMed]
- Ju, B.; Zhang, Q.; Ge, J.; Wang, R.; Sun, J.; Ge, X.; Yu, J.; Shan, S.; Zhou, B.; Song, S.; et al. Human Neutralizing Antibodies Elicited by SARS-CoV-2 Infection. Nature 2020, 584, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Duyvesteyn, H.M.E.; Chen, C.-P.; Huang, C.-G.; Chen, T.-H.; Shih, S.-R.; Lin, Y.-C.; Cheng, C.-Y.; Cheng, S.-H.; Huang, Y.-C.; et al. Structural Basis for the Neutralization of SARS-CoV-2 by an Antibody from a Convalescent Patient. Nat. Struct. Mol. Biol. 2020, 27, 950–958. [Google Scholar] [CrossRef]
- Du, S.; Cao, Y.; Zhu, Q.; Yu, P.; Qi, F.; Wang, G.; Du, X.; Bao, L.; Deng, W.; Zhu, H.; et al. Structurally Resolved SARS-CoV-2 Antibody Shows High Efficacy in Severely Infected Hamsters and Provides a Potent Cocktail Pairing Strategy. Cell 2020, 183, 1013.e13–1023.e13. [Google Scholar] [CrossRef]
- Rujas, E.; Kucharska, I.; Tan, Y.Z.; Benlekbir, S.; Cui, H.; Zhao, T.; Wasney, G.A.; Budylowski, P.; Guvenc, F.; Newton, J.C.; et al. Multivalency transforms SARS-CoV-2 antibodies into ultrapotent neutralizers. Nat. Commun. 2021, 12, 3611. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, L.; Park, Y.-J.; Tortorici, M.A.; Czudnochowski, N.; Walls, A.C.; Beltramello, M.; Silacci-Fregni, C.; Pinto, D.; Rosen, L.E.; Bowen, J.E.; et al. Mapping Neutralizing and Immunodominant Sites on the SARS-CoV-2 Spike Receptor-Binding Domain by Structure-Guided High-Resolution Serology. Cell 2020, 183, 1024.e21–1042.e21. [Google Scholar] [CrossRef] [PubMed]
- Tortorici, M.A.; Beltramello, M.; Lempp, F.A.; Pinto, D.; Dang, H.V.; Rosen, L.E.; McCallum, M.; Bowen, J.; Minola, A.; Jaconi, S.; et al. Ultrapotent Human Antibodies Protect against SARS-CoV-2 Challenge via Multiple Mechanisms. Science 2020, 370, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.C.; Yuan, M.; Liu, H.; Lee, C.-C.D.; Zhu, X.; Bangaru, S.; Torres, J.L.; Caniels, T.G.; Brouwer, P.J.M.; van Gils, M.J.; et al. An Alternative Binding Mode of IGHV3-53 Antibodies to the SARS-CoV-2 Receptor Binding Domain. Cell Rep. 2020, 33, 108274. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Deng, Y.-Q.; Ye, Q.; Cao, L.; Sun, C.-Y.; Fan, C.; Huang, W.; Sun, S.; Sun, Y.; Zhu, L.; et al. Structural Basis for Neutralization of SARS-CoV-2 and SARS-CoV by a Potent Therapeutic Antibody. Science 2020, 369, 1505–1509. [Google Scholar] [CrossRef]
- Liu, H.; Wu, N.C.; Yuan, M.; Bangaru, S.; Torres, J.L.; Caniels, T.G.; van Schooten, J.; Zhu, X.; Lee, C.-C.D.; Brouwer, P.J.M.; et al. Cross-Neutralization of a SARS-CoV-2 Antibody to a Functionally Conserved Site Is Mediated by Avidity. bioRxiv 2020. [Google Scholar] [CrossRef]
- Huo, J.; Zhao, Y.; Ren, J.; Zhou, D.; Duyvesteyn, H.M.E.; Ginn, H.M.; Carrique, L.; Malinauskas, T.; Ruza, R.R.; Shah, P.N.M.; et al. Neutralization of SARS-CoV-2 by Destruction of the Prefusion Spike. Cell Host Microbe 2020, 28, 445.e6–454.e6. [Google Scholar] [CrossRef]
- Wrobel, A.G.; Benton, D.J.; Hussain, S.; Harvey, R.; Martin, S.R.; Roustan, C.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Antibody-Mediated Disruption of the SARS-CoV-2 Spike Glycoprotein. Nat. Commun. 2020, 11, 5337. [Google Scholar] [CrossRef]
- Shi, R.; Shan, C.; Duan, X.; Chen, Z.; Liu, P.; Song, J.; Song, T.; Bi, X.; Han, C.; Wu, L.; et al. A Human Neutralizing Antibody Targets the Receptor-Binding Site of SARS-CoV-2. Nature 2020, 584, 120–124. [Google Scholar] [CrossRef]
- Yuan, M.; Liu, H.; Wu, N.C.; Lee, C.-C.D.; Zhu, X.; Zhao, F.; Huang, D.; Yu, W.; Hua, Y.; Tien, H.; et al. Structural Basis of a Shared Antibody Response to SARS-CoV-2. Science 2020, 369, 1119–1123. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, F.; Shen, C.; Peng, W.; Li, D.; Zhao, C.; Li, Z.; Li, S.; Bi, Y.; Yang, Y.; et al. A Noncompeting Pair of Human Neutralizing Antibodies Block COVID-19 Virus Binding to Its Receptor ACE2. Science 2020, 368, 1274–1278. [Google Scholar] [CrossRef]
- Barnes, C.O.; Jette, C.A.; Abernathy, M.E.; Dam, K.-M.A.; Esswein, S.R.; Gristick, H.B.; Malyutin, A.G.; Sharaf, N.G.; Huey-Tubman, K.E.; Lee, Y.E.; et al. SARS-CoV-2 Neutralizing Antibody Structures Inform Therapeutic Strategies. Nature 2020, 588, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Vangone, A.; Bonvin, A.M. Contacts-Based Prediction of Binding Affinity in Protein-Protein Complexes. Elife 2015, 4, e07454. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A Web Server for Predicting the Binding Affinity of Protein-Protein Complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef] [PubMed]
- Dehouck, Y.; Kwasigroch, J.M.; Rooman, M.; Gilis, D. BeAtMuSiC: Prediction of Changes in Protein-Protein Binding Affinity on Mutations. Nucleic Acids Res. 2013, 41, W333–W339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, C.; Xue, L.C.; Roel-Touris, J.; Bonvin, A.M.J.J. Finding the ΔΔ G Spot: Are Predictors of Binding Affinity Changes upon Mutations in Protein–Protein Interactions Ready for It? Wiley Interdiscip. Rev. Comput. Mol. Sci. 2019, 9, e1410. [Google Scholar] [CrossRef] [Green Version]
- GISAID—Initiative. Available online: https://www.gisaid.org/ (accessed on 11 March 2021).
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Bakan, A.; Dutta, A.; Mao, W.; Liu, Y.; Chennubhotla, C.; Lezon, T.R.; Bahar, I. Evol and ProDy for Bridging Protein Sequence Evolution and Structural Dynamics. Bioinformatics 2014, 30, 2681–2683. [Google Scholar] [CrossRef] [Green Version]
- Dunn, S.D.; Wahl, L.M.; Gloor, G.B. Mutual Information without the Influence of Phylogeny or Entropy Dramatically Improves Residue Contact Prediction. Bioinformatics 2008, 24, 333–340. [Google Scholar] [CrossRef] [Green Version]
- Woo, H.; Park, S.-J.; Choi, Y.K.; Park, T.; Tanveer, M.; Cao, Y.; Kern, N.R.; Lee, J.; Yeom, M.S.; Croll, T.I.; et al. Developing a Fully Glycosylated Full-Length SARS-CoV-2 Spike Protein Model in a Viral Membrane. J. Phys. Chem. B 2020, 124, 7128–7137. [Google Scholar] [CrossRef]
- CHARMM-GUI. Available online: https://charmm-gui.org/?doc=archive&lib=covid19 (accessed on 25 January 2022).
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Tortorici, M.A.; Veesler, D. Structural Insights into Coronavirus Entry. Adv. Virus Res. 2019, 105, 93–116. [Google Scholar] [PubMed]
- Grant, O.C.; Montgomery, D.; Ito, K.; Woods, R.J. Analysis of the SARS-CoV-2 Spike Protein Glycan Shield Reveals Implications for Immune Recognition. Sci. Rep. 2020, 10, 14991. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.I.; MacGowan, S.A.; Kutuzov, M.A.; Dushek, O.; Barton, G.J.; van der Merwe, P.A. Effects of Common Mutations in the SARS-CoV-2 Spike RBD and Its Ligand, the Human ACE2 Receptor on Binding Affinity and Kinetics. eLife 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Wu, N.C.; Zhu, X.; Lee, C.-C.D.; So, R.T.Y.; Lv, H.; Mok, C.K.P.; Wilson, I.A. A Highly Conserved Cryptic Epitope in the Receptor Binding Domains of SARS-CoV-2 and SARS-CoV. Science 2020, 368, 630–633. [Google Scholar] [CrossRef] [Green Version]
- Phillips, P.C. Epistasis--the Essential Role of Gene Interactions in the Structure and Evolution of Genetic Systems. Nat. Rev. Genet. 2008, 9, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Greaney, A.J.; Starr, T.N.; Barnes, C.O.; Weisblum, Y.; Schmidt, F.; Caskey, M.; Gaebler, C.; Cho, A.; Agudelo, M.; Finkin, S.; et al. Mapping Mutations to the SARS-CoV-2 RBD That Escape Binding by Different Classes of Antibodies. Nat. Commun. 2021, 12, 4196. [Google Scholar] [CrossRef]
- Morcos, F.; Onuchic, J.N. The Role of Coevolutionary Signatures in Protein Interaction Dynamics, Complex Inference, Molecular Recognition, and Mutational Landscapes. Curr. Opin. Struct. Biol. 2019, 56, 179–186. [Google Scholar] [CrossRef]
- Wu, N.C.; Xie, J.; Zheng, T.; Nycholat, C.M.; Grande, G.; Paulson, J.C.; Lerner, R.A.; Wilson, I.A. Diversity of Functionally Permissive Sequences in the Receptor-Binding Site of Influenza Hemagglutinin. Cell Host Microbe 2017, 22, 247–248. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182, 1284.e9–1294.e9. [Google Scholar] [CrossRef]
- Yi, C.; Sun, X.; Ye, J.; Ding, L.; Liu, M.; Yang, Z.; Lu, X.; Zhang, Y.; Ma, L.; Gu, W.; et al. Key Residues of the Receptor Binding Motif in the Spike Protein of SARS-CoV-2 That Interact with ACE2 and Neutralizing Antibodies. Cell. Mol. Immunol. 2020, 17, 621–630. [Google Scholar] [CrossRef]
- Ku, Z.; Xie, X.; Davidson, E.; Ye, X.; Su, H.; Menachery, V.D.; Li, Y.; Yuan, Z.; Zhang, X.; Muruato, A.E.; et al. Molecular Determinants and Mechanism for Antibody Cocktail Preventing SARS-CoV-2 Escape. Nat. Commun. 2021, 12, 469. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. Could New COVID Variants Undermine Vaccines? Labs Scramble to Find Out. Nature 2021, 589, 177–178. [Google Scholar] [CrossRef] [PubMed]
- Abdelrahman, Z.; Li, M.; Wang, X. Comparative Review of SARS-CoV-2, SARS-CoV, MERS-CoV, and Influenza A Respiratory Viruses. Front. Immunol. 2020, 11, 552909. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Li, C.; Huang, A.; Xia, S.; Lu, S.; Shi, Z.; Lu, L.; Jiang, S.; Yang, Z.; Wu, Y.; et al. Potent Binding of 2019 Novel Coronavirus Spike Protein by a SARS Coronavirus-Specific Human Monoclonal Antibody. Emerg. Microbes Infect. 2020, 9, 382–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planas, D.; Saunders, N.; Maes, P.; Guivel-Benhassine, F.; Planchais, C.; Buchrieser, J.; Bolland, W.-H.; Porrot, F.; Staropoli, I.; Lemoine, F.; et al. Considerable Escape of SARS-CoV-2 Omicron to Antibody Neutralization. Nature 2021, 580, 7803. [Google Scholar] [CrossRef]
- Wolter, N.; Jassat, W.; Walaza, S.; Welch, R.; Moultrie, H.; Groome, M.; Amoako, D.G.; Everatt, J.; Bhiman, J.N.; Scheepers, C.; et al. Early Assessment of the Clinical Severity of the SARS-CoV-2 Omicron Variant in South Africa. bioRxiv 2021. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
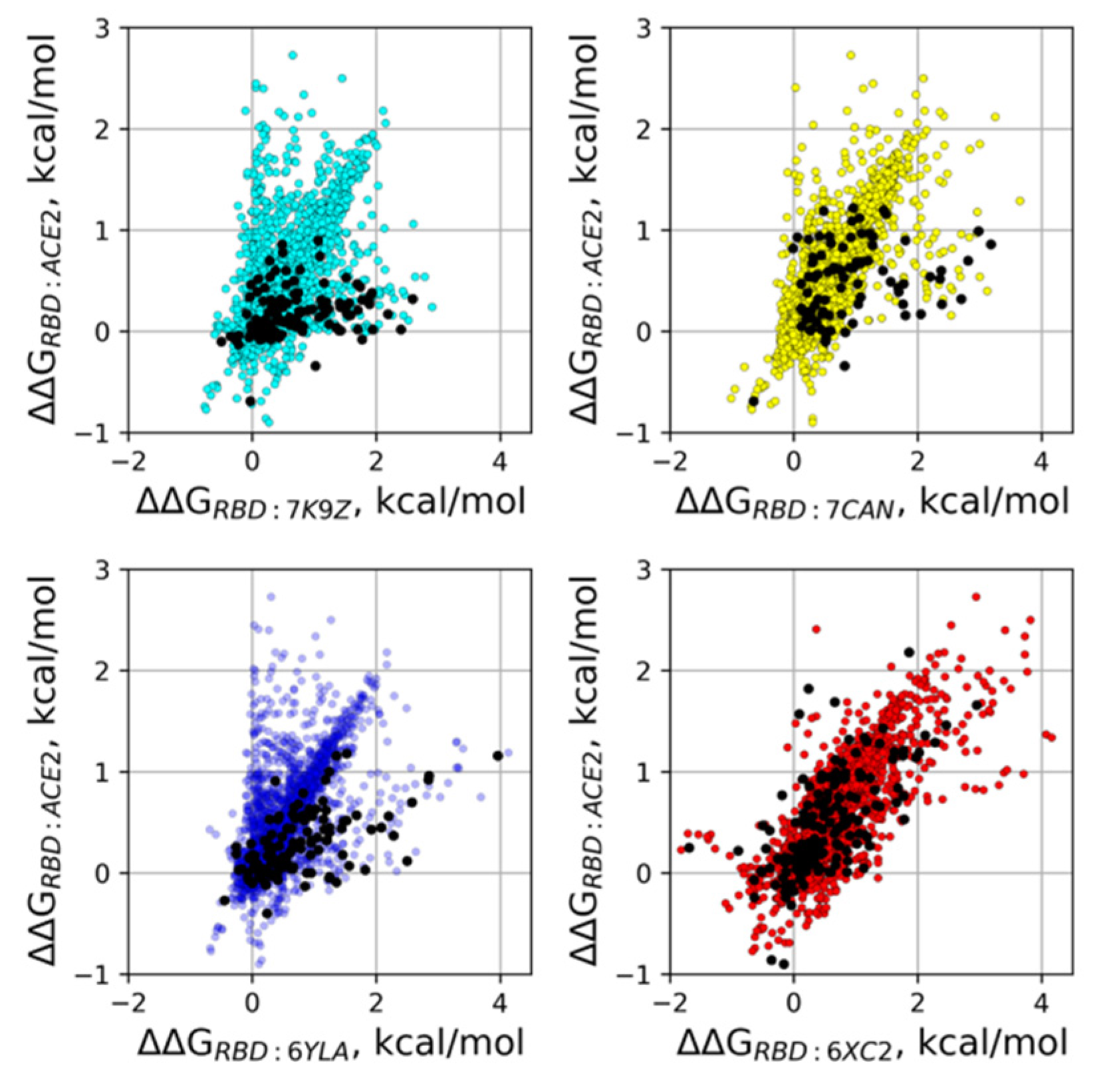{kind=link}
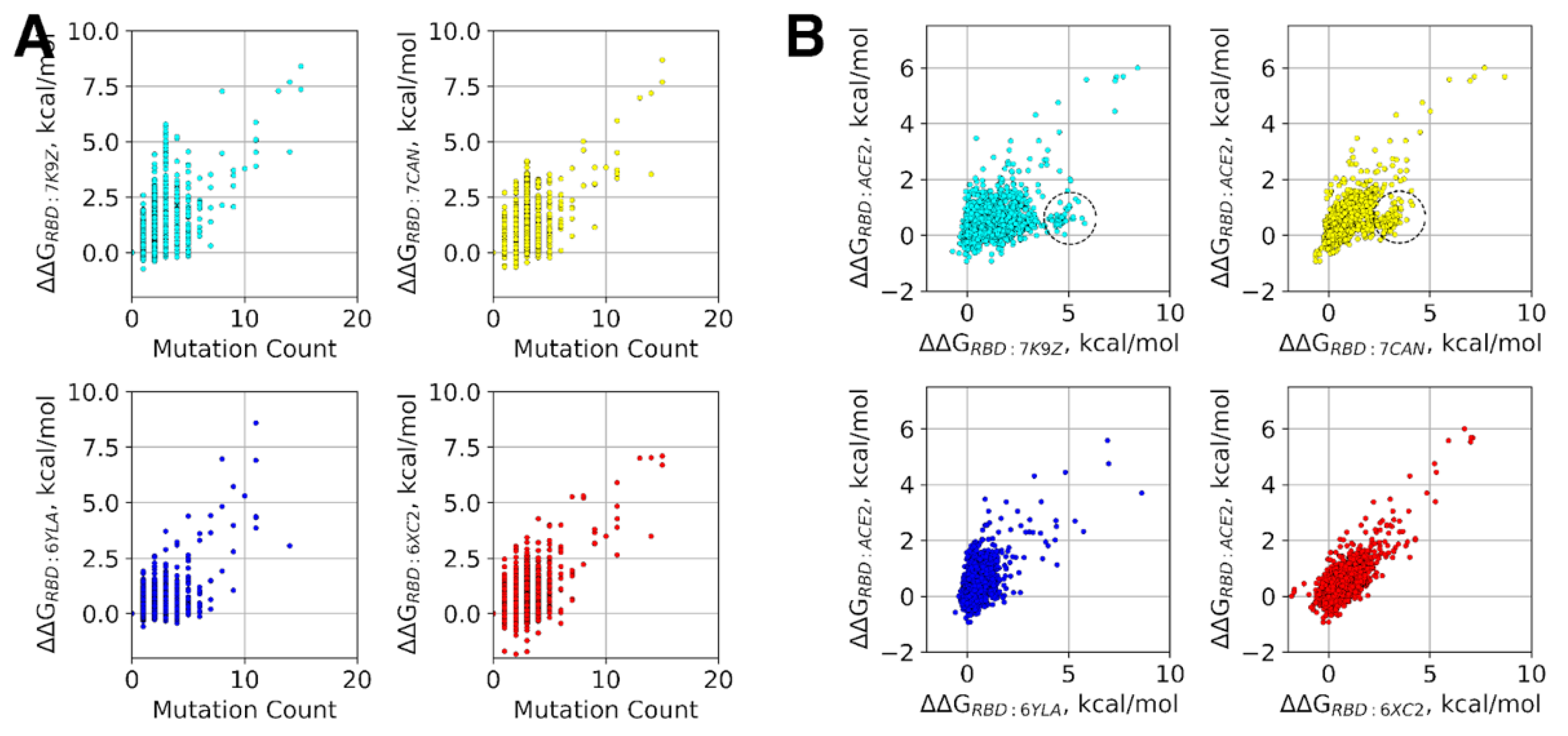{kind=link}
| Cluster | Complex | ΔG, kcal/mol | No. Contacts | Mean Interface Variability, Bit | Antibody Type/Name | PDB Chains | Ref. | ||
|---|---|---|---|---|---|---|---|---|---|
| RBD | H | L | |||||||
| 1 | 6XDG_CA | −10.7 | 49 | 0.09 | REGN10987 antibody Fab | E | C | A | [25] |
| 1 | 6ZBP_B | −9.6 | 53 | 0.105 | H11-H4 nanobody | A | B | - | |
| 1 | 6XKP_HL | −9.3 | 52 | 0.092 | neutralizing antibody CV07-270 | A | H | L | [26] |
| 1 | 7BWJ_HL | −9.6 | 56 | 0.116 | P2B-2F6 Fab | E | H | L | [27] |
| 1 | 6ZCZ_F | −9.6 | 55 | 0.097 | nanobody H11-H4 | E | F | [28] | |
| 1 | 7C8V_A | −9.9 | 54 | 0.145 | synthetic nanobody SR4 | B | A | - | |
| 1 | 7CHC_AB | −9.5 | 58 | 0.106 | BD-368-2 Fab | R | A | B | [29] |
| 1 | 7K9Z_HL | −11.5 | 75 | 0.081 | Fab fragment neutralizing antibody 52 | E | H | L | [30] |
| 1 | 7JX3_AB | −9.8 | 63 | 0.064 | Fab domain of monoclonal antibody S309 | R | A | B | [31] |
| 2 | 7JX3_CD | −11.2 | 82 | 0.09 | Fab domain of monoclonal antibody S2H14 | R | C | D | [31] |
| 2 | 7C8W_A | −11.2 | 79 | 0.091 | synthetic nanobody MR17 | B | A | - | |
| 2 | 6XDG_BD | −10.2 | 69 | 0.102 | REGN10933 antibody Fab | E | B | D | [25] |
| 2 | 6XKQ_HL | −9.8 | 70 | 0.115 | neutralizing antibody CV07-250 | A | H | L | [26] |
| 2 | 7JV2_HL | −10.3 | 56 | 0.094 | S2H13 neutralizing antibody Fab fragment | A | H | L | [31] |
| 2 | 7K45_HL | −10.9 | 66 | 0.1 | S2E12 neutralizing antibody Fab | B | H | L | [32] |
| 2 | 7K9Z_AB | −9.7 | 56 | 0.132 | Fab fragment neutralizing antibody 298 | E | A | B | [30] |
| 2 | 7CAN_A | −11.5 | 77 | 0.088 | synthetic nanobody MR17-K99Y | B | A | - | |
| 2 | 7JMP_HL | −8.3 | 52 | 0.101 | neutralizing antibody COVA2-39 | A | H | L | [33] |
| 3 | 6ZCZ_HL | −12.3 | 80 | 0.038 | EY6A Fab | E | H | L | [28] |
| 3 | 7CAH_ED | −12.6 | 86 | 0.045 | H014 Fab | A | E | D | [34] |
| 3 | 7JMW_HL | −10.7 | 58 | 0.039 | cross-neutralizing antibody COVA1-16 Fab | A | H | L | [35] |
| 3 | 6YLA_HL | −14.9 | 99 | 0.04 | CR3022 Fab | E | H | L | [36] |
| 3 | 7JX3_HL | −12.4 | 83 | 0.042 | Fab domain of monoclonal antibody S304 | R | H | L | [31] |
| 3 | 7JVA_HL | −10.2 | 72 | 0.049 | S2A4 neutralizing antibody Fab fragment | A | H | L | [31] |
| 3 | 7A5S_HL | −14 | 102 | 0.047 | CR3022 Fab | A | H | L | [37] |
| 4 | 7C01_HL | −13.4 | 112 | 0.094 | neutralizing antibody CB6 | A | H | L | [38] |
| 4 | 7CH5_HL | −12.5 | 97 | 0.101 | BD-629 Fab | R | H | L | [29] |
| 4 | 6XC2_HL | −16.4 | 137 | 0.083 | neutralizing antibody CC12.1 | A | H | L | [39] |
| 4 | 7CH4_HL | −15.9 | 114 | 0.09 | BD-604 Fab | R | H | L | [29] |
| 4 | 7BZ5_HL | −14.6 | 121 | 0.088 | neutralizing antibody B38 | A | H | L | [40] |
| 4 | 7JMO_HL | −12.9 | 108 | 0.086 | neutralizing antibody COVA2-04 | A | H | L | [33] |
| 4 | 7CHB_HL | −13.5 | 110 | 0.087 | BD-236 Fab | R | H | L | [29] |
| 4 | 6XC4_HL | −14 | 99 | 0.104 | neutralizing antibody CC12.3 | A | H | L | [39] |
| 4 | 7CHC_HL | −12 | 95 | 0.101 | BD-629 Fab | R | H | L | [29] |
| 4 | 7K8M_AB | −13.5 | 110 | 0.093 | Fab fragment neutralizing antibody C102 | E | A | B | [41] |
| - | 6M17_EB | −11.4 | 66 | 0.101 | ACE2 receptor | E | B | [24] | |
| Variant(s) | RBD Mutations |
|---|---|
| B.1.1.7 (α) | N501Y |
| B.1.351 (β) | K417N, E484K, N501Y |
| P.1 (γ) | K417T, E484K, N501Y |
| B.1.617.2 (δ) | L452R, T478K |
| B.1.617.2+ (δ+) | L452R, K417N, T478K |
| B.1.427/B.1.429 (ε) | L452R |
| P.2, B.1.525, B.1.526 (ζ, η, ι) | E484K |
| P.3 (θ) | E484K, N501Y |
| B.1.617.1 (κ) | L452R, E484Q |
| C.37 (λ) | L452Q, F490S |
| B.1.621 (μ) | R346K, E484K, N501Y |
| B.1.1.529 (ο) | G339D, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493R, G496S, Q498R, N501Y, Y505H |
| ID | Origin | No. Mutations | ΔΔG, kcal/mol | ||||
|---|---|---|---|---|---|---|---|
| RBD:7K9Z | RBD:7CAN | RBD:6YLA | RBD:6XC2 | RBD:ACE2 | |||
| EPI_ISL_410541 | pangolin/Guangxi | 25 | 4.68 | 9.52 | 3.41 | 8.21 | 6.93 |
| EPI_ISL_410538 | pangolin/Guangxi | 24 | 3.78 | 8.46 | 2.77 | 7.54 | 5.81 |
| EPI_ISL_410539 | pangolin/Guangxi | 23 | 3.21 | 8.2 | 2.54 | 7.41 | 5.58 |
| EPI_ISL_402131 | bat/Yunnan | 20 | 3.46 | 7.37 | 2.08 | 7.78 | 5.6 |
| EPI_ISL_568499 | human/Iran | 8 | 7.27 | 5.02 | 4.83 | 5.31 | 4.44 |
| EPI_ISL_568500 | human/Iran | 11 | 3.9 | 3.35 | 4.37 | 2.65 | 2.71 |
| B.1.351 (β) | 3 | 0.3 | 0.48 | 0.13 | 0.8 | 0.29 | |
| B.1.617.2+ (δ+) | 3 | 1.49 | 0.87 | 0.55 | 1.39 | 0.58 | |
| B.1.621 (μ) | 3 | 0.5 | 0.21 | −0.19 | −0.23 | −0.16 | |
| B.1.1.529 (ο) | 15 | 1.44 | 4.2 | 1.08 | 5.59 | 4.92 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bozdaganyan, M.E.; Shaitan, K.V.; Kirpichnikov, M.P.; Sokolova, O.S.; Orekhov, P.S. Computational Analysis of Mutations in the Receptor-Binding Domain of SARS-CoV-2 Spike and Their Effects on Antibody Binding. Viruses 2022, 14, 295. https://doi.org/10.3390/v14020295
Bozdaganyan ME, Shaitan KV, Kirpichnikov MP, Sokolova OS, Orekhov PS. Computational Analysis of Mutations in the Receptor-Binding Domain of SARS-CoV-2 Spike and Their Effects on Antibody Binding. Viruses. 2022; 14(2):295. https://doi.org/10.3390/v14020295
Chicago/Turabian StyleBozdaganyan, Marine E., Konstantin V. Shaitan, Mikhail P. Kirpichnikov, Olga S. Sokolova, and Philipp S. Orekhov. 2022. "Computational Analysis of Mutations in the Receptor-Binding Domain of SARS-CoV-2 Spike and Their Effects on Antibody Binding" Viruses 14, no. 2: 295. https://doi.org/10.3390/v14020295
APA StyleBozdaganyan, M. E., Shaitan, K. V., Kirpichnikov, M. P., Sokolova, O. S., & Orekhov, P. S. (2022). Computational Analysis of Mutations in the Receptor-Binding Domain of SARS-CoV-2 Spike and Their Effects on Antibody Binding. Viruses, 14(2), 295. https://doi.org/10.3390/v14020295