
Academ Virus, a Novel Hantavirus in the Siberian Mole (Talpa altaica) from Russia

 , ,
, ,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
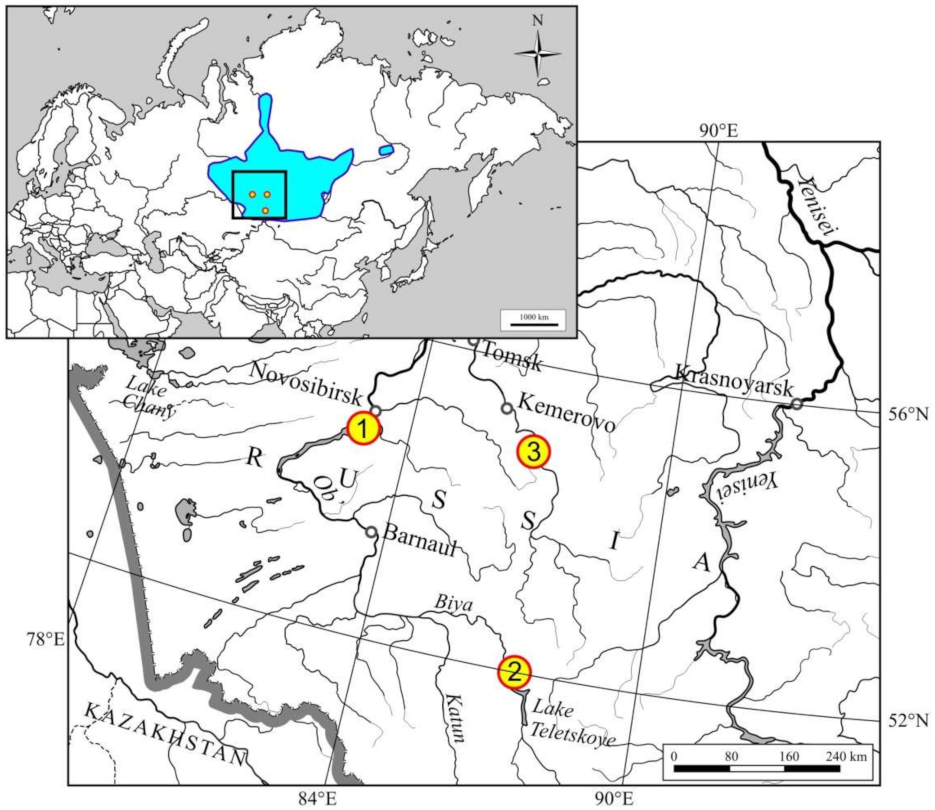
2.1. Trapping and Sample Collection
2.2. RNA Extraction and RT-PCR Analysis
2.3. Genetic and Phylogenetic Analysis
3. Results
3.1. Genetic Analysis
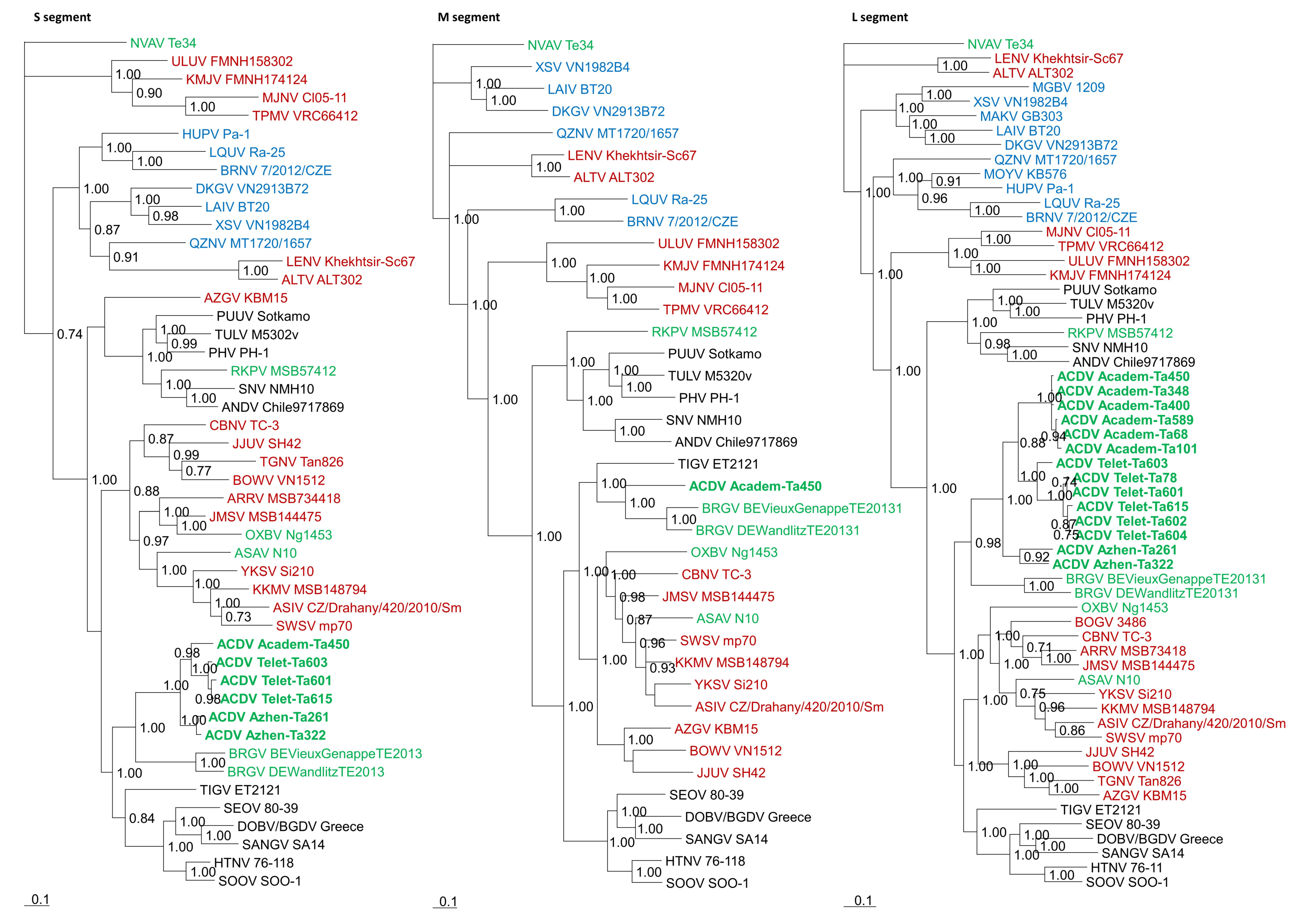
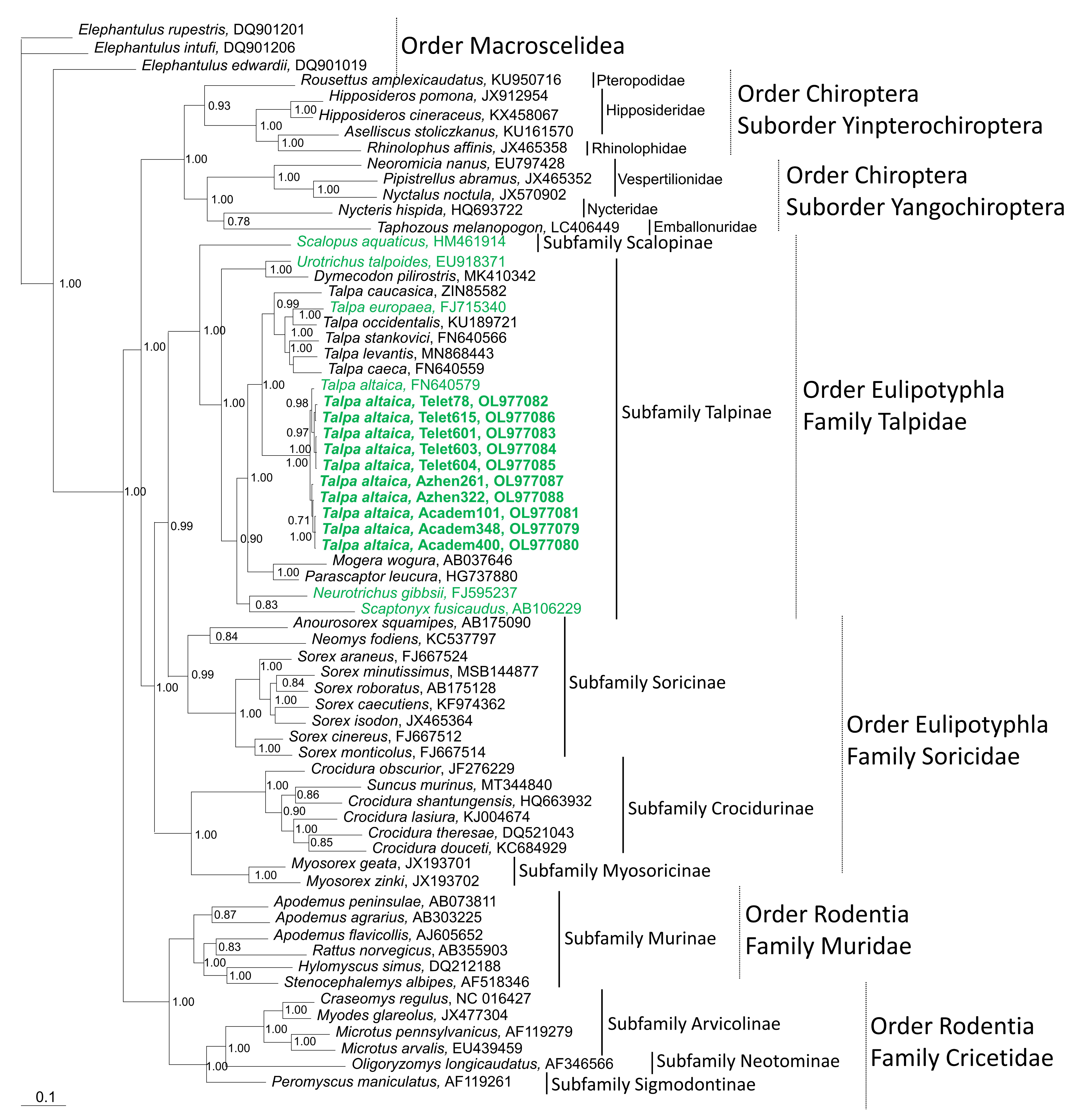
3.2. Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yanagihara, R.; Gu, S.H.; Arai, S.; Kang, H.J.; Song, J.-W. Hantaviruses: Rediscovery and new beginnings. Virus Res. 2014, 187, 6–14. [Google Scholar] [CrossRef] [Green Version]
- Arai, S.; Yanagihara, R. Genetic diversity and geographic distribution of bat-borne hantaviruses. Curr. Issues Mol. Biol. 2020, 39, 1–28. [Google Scholar] [CrossRef]
- Jonsson, C.B.; Figueiredo, L.T.; Vapalahti, O. A global perspective on hantavirus ecology, epidemiology, and disease. Clin. Microbiol. Rev. 2010, 23, 412–444. [Google Scholar] [CrossRef] [Green Version]
- Clement, J.; Maes, P.; Lagron, K.; Ranst, M.; Lameire, N. A unifying hypothesis and a single name for a complex globally emerging infection: Hantavirus disease. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Heinemann, P.; Tia, M.; Alabi, A.; Anon, J.C.; Auste, B.; Essbauer, S.; Gnionsahe, A.; Kigninlman, H.; Klempa, B.; Kraef, C.; et al. Human infections by non-rodent-associated hantaviruses in Africa. J. Infect. Dis. 2016, 214, 1507–1511. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Shimizu, K.; Nishigami, K.; Tsuda, Y.; Sarathukumara, Y.; Muthusinghe, D.S.; Gamage, C.D.; Granathne, L.; Lokupathirage, S.M.W.; Nanayakkara, N.; et al. Serological methods for detection of infection with shrew-borne hantaviruses: Thottapalayam, Seewis, Altai, and Asama viruses. Arch. Virol. 2021, 166, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Maes, P.; Adkins, S.; Alkhovsky, S.V.; Avšič-Županc, T.; Ballinger, M.J.; Bente, D.A.; Beer, M.; Bergeron, E.; Blair, C.D.; Briese, T.; et al. Taxonomy of the order Bunyavirales: Second update 2018. Arch. Virol. 2019, 164, 927–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vapalahti, O.; Lundkvist, A.; Fedorov, V.; Conroy, C.J.; Hirvonen, S.; Plyusnina, A.; Nemirov, K.; Fredga, K.; Cook, J.A.; Niemimaa, J.; et al. Isolation and characterization of a hantavirus from Lemmus sibiricus: Evidence for host switch during hantavirus evolution. J. Virol. 1999, 73, 5586–5592. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.J.; Bennett, S.N.; Dizney, L.; Sumibcay, L.; Arai, S.; Ruedas, L.A.; Song, J.W.; Yanagihara, R. Host switch during evolution of a genetically distinct hantavirus in the American shrew mole (Neurotrichus gibbsii). Virology 2009, 388, 8–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, J.; Sironen, T.; Voutilainen, L.; Hepojoki, S.; Niemimaa, J.; Isoviita, V.M.; Vaheri, A.; Henttonen, H.; Vapalahti, O. Han-taviruses in Finnish soricomorphs: Evidence for two distinct hantaviruses carried by Sorex araneus suggesting ancient host-switch. Infect. Genet. Evol. 2014, 27, 51–61. [Google Scholar] [CrossRef]
- Kang, H.J.; Gu, S.H.; Yashina, L.N.; Cook, J.A.; Yanagihara, R. Highly divergent genetic variants of soricid-borne Altai virus (Hantaviridae) in Eurasia suggest ancient host-switching events. Viruses 2019, 11, 857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liphardt, S.W.; Kang, H.J.; Dizney, L.J.; Ruedas, L.A.; Cook, J.A.; Yanagihara, R. Complex history of codiversification and host switching of a newfound soricid-borne orthohantavirus in North America. Viruses 2019, 11, 637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guterres, A.; de Oliveira, R.C.; Fernandes, J.; de Lemos, E.R.S. The mystery of the phylogeographic structural pattern in rodent-borne hantaviruses. Mol. Phylogenet. Evol. 2019, 136, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Schmaljohn, A.L.; Anderson, K.; Schmaljohn, C.S. Complete nucleotide sequences of the M and S segments of two hantavirus isolates from California: Evidence for reassortment in nature among viruses related to hantavirus pulmonary syndrome. Virology 1995, 206, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Razzauti, M.; Plyusnina, A.; Sironen, T.; Henttonen, H.; Plyusnin, A. Analysis of Puumala hantavirus in a bank vole population in northern Finland: Evidence for co-circulation of two genetic lineages and frequent reassortment between strains. J. Gen. Virol. 2009, 90, 1923–1931. [Google Scholar] [CrossRef] [Green Version]
- Klempa, B. Reassortment events in the evolution of hantaviruses. Virus Genes 2018, 54, 638–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laenen, L.; Vergote, V.; Kafetzopoulou, L.E.; Wawina, T.B.; Vassou, D.; Cook, J.A.; Hugot, J.P.; Deboutte, W.; Kang, H.J.; Witkowski, P.T.; et al. A novel hantavirus of the European mole, Bruges virus, is involved in frequent Nova virus coinfections. Genome Biol. Evol. 2018, 10, 45–55. [Google Scholar] [CrossRef]
- Liphardt, S.W.; Kang, H.J.; Arai, S.; Gu, S.H.; Cook, J.A.; Yanagihara, R. Reassortment between divergent strains of Camp Ripley virus (Hantaviridae) in the northern short-tailed shrew (Blarina brevicauda). Front. Cell Infect. Microbiol. 2020, 10, 460. [Google Scholar] [CrossRef]
- Kang, H.J.; Gu, S.H.; Cook, J.A.; Yanagihara, R. Dahonggou Creek virus, a divergent lineage of hantavirus harbored by the long-tailed mole (Scaptonyx fusicaudus). Trop. Med. Health. 2016, 44, 16. [Google Scholar] [CrossRef] [Green Version]
- Arai, S.; Ohdachi, S.D.; Asakawa, M.; Kang, H.J.; Mocz, G.; Arikawa, J.; Okabe, N.; Yanagihara, R. Molecular phylogeny of a newfound hantavirus in the Japanese shrew mole (Urotrichus talpoides). Proc. Natl. Acad. Sci. USA 2008, 105, 16296–16301. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.J.; Bennett, S.N.; Sumibcay, L.; Arai, S.; Hope, A.G.; Mocz, G.; Song, J.-W.; Cook, J.A.; Yanagihara, R. Evolutionary insights from a genetically divergent hantavirus harbored by the European common mole (Talpa europaea). PLoS ONE 2009, 4, e6149. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Bennett, S.N.; Hope, A.G.; Cook, J.A.; Yanagihara, R. Shared ancestry between a newfound mole-borne hantavirus and hantaviruses harbored by cricetid rodents. J. Virol. 2011, 85, 7496–7503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, S.H.; Hejduk, J.; Markowski, J.; Kang, H.J.; Markowski, M.; Połatynska, M.; Sikorska, B.; Liberski, P.P.; Yanagihara, R. Co-circulation of genetically distinct soricid- and talpid-borne hantaviruses in Poland. Infect. Genet. Evol. 2014, 28, 296–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klempa, B.; Fichet-Calvet, E.; Lecompte, E.; Auste, B.; Aniskin, V.; Meisel, H.; Barrier, P.; Koivogue, L.; Meulen, J.; Krüger, D.H. Novel hantavirus sequences in shrew, Guinea. Emerg. Infect. Dis. 2007, 13, 520–522. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Zardoya, R.; Telford, M.J. TranslatorX: Multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010, 38, W7–W13. [Google Scholar] [CrossRef] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence align-ment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [Green Version]
- Posada, D.; Crandall, K.A. MODELTEST: Testing the model of DNA substitution. Bioinformatics 1998, 14, 817–818. [Google Scholar] [CrossRef] [Green Version]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Yashina, L.N.; Kartashov, M.Y.; Wang, W.; Li, K.; Zdanovskaya, N.I.; Ivanov, L.I.; Zhang, Y.Z. Co-circulation of distinct shrew-borne hantaviruses in the far east of Russia. Virus Res. 2019, 272, 197717. [Google Scholar] [CrossRef] [PubMed]
- Yashina, L.N.; Abramov, S.A.; Zhigalin, A.V.; Smetannikova, N.A.; Dupal, T.A.; Krivopalov, A.V.; Kikuchi, F.; Senoo, K.; Arai, S.; Mizutani, T.; et al. Geographic distribution and phylogeny of soricine shrew-borne Seewis virus and Altai virus in Russia. Viruses 2021, 13, 1286. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Z. Discovery of hantaviruses in bats and insectivores and the evolution of the genus Hantavirus. Virus Res. 2014, 187, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Kallio, E.R.; Klingström, J.; Gustafsson, E.; Manni, T.; Vaheri, A.; Henttonen, H.; Vapalahti, O.; Lundkvist, Å. Prolonged survival of Puumala hantavirus outside the host: Evidence for indirect transmission via the environment. J. Gen. Virol. 2006, 87, 2127–2134. [Google Scholar] [CrossRef]
- Hewitt, G.M. Some genetic consequences of ice ages, and their role in divergence and speciation. Biol. J. Lin. Soc. 1996, 58, 247–276. [Google Scholar] [CrossRef]
- Řičánková, V.P.; Robovský, J.; Riegert, J. Ecological structure of Recent and Last Glacial mammalian faunas in northern Eurasia: The case of Altai-Sayan refugium. PLoS ONE 2014, 9, e85056. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Number Hantavirus Positive/Tested | GenBank No. | |||||
|---|---|---|---|---|---|---|
| Capture Site | Year | Moles | Virus Strain | S | M | L |
| Novosibirsk Oblast, Academgorodok | 2017 | 1/2 | Academ-Ta450 | MK340905 | OL871119 | MH784614 |
| 2019 | 3/3 | Academ-Ta348 | - | - | MZ062416 | |
| Academ-Ta400 | - | - | MZ062417 | |||
| Academ-Ta589 | - | - | MZ062418 | |||
| 2021 | 2/2 | Academ-Ta68 | - | - | OL871122 | |
| Academ-Ta101 | - | - | OL871123 | |||
| Altai Republic, Teletskoye | 2018 | 1/1 | Telet-Ta78 | MZ062419 | ||
| 2020 | 5/6 | Telet-Ta601 | MZ062425 | - | MZ062420 | |
| Telet-Ta602 | - | - | MZ062421 | |||
| Telet-Ta603 | MZ062426 | - | MZ062422 | |||
| Telet-Ta604 | - | - | MZ062423 | |||
| Telet-Ta615 | MZ062427 | - | MZ062424 | |||
| Kemerovo Oblast, Azhendarovo | 2021 | 2/4 | Azhen-Ta261 | OL871120 | - | OL871124 |
| Azhen-Ta322 | OL871121 | - | OL871125 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yashina, L.N.; Panov, V.V.; Abramov, S.A.; Smetannikova, N.A.; Luchnikova, E.M.; Dupal, T.A.; Krivopalov, A.V.; Arai, S.; Yanagihara, R. Academ Virus, a Novel Hantavirus in the Siberian Mole (Talpa altaica) from Russia. Viruses 2022, 14, 309. https://doi.org/10.3390/v14020309
Yashina LN, Panov VV, Abramov SA, Smetannikova NA, Luchnikova EM, Dupal TA, Krivopalov AV, Arai S, Yanagihara R. Academ Virus, a Novel Hantavirus in the Siberian Mole (Talpa altaica) from Russia. Viruses. 2022; 14(2):309. https://doi.org/10.3390/v14020309
Chicago/Turabian StyleYashina, Liudmila N., Victor V. Panov, Sergey A. Abramov, Natalia A. Smetannikova, Ekaterina M. Luchnikova, Tamara A. Dupal, Anton V. Krivopalov, Satoru Arai, and Richard Yanagihara. 2022. "Academ Virus, a Novel Hantavirus in the Siberian Mole (Talpa altaica) from Russia" Viruses 14, no. 2: 309. https://doi.org/10.3390/v14020309
APA StyleYashina, L. N., Panov, V. V., Abramov, S. A., Smetannikova, N. A., Luchnikova, E. M., Dupal, T. A., Krivopalov, A. V., Arai, S., & Yanagihara, R. (2022). Academ Virus, a Novel Hantavirus in the Siberian Mole (Talpa altaica) from Russia. Viruses, 14(2), 309. https://doi.org/10.3390/v14020309