2. Materials and Methods
2.1. General Methods
Unless otherwise stated, reagents and solvents were purchased from commercial sources (Sigma Aldrich, St Louis, MO, USA; Acros Organics, Carlsbad, CA, USA; Merck Chemicals, Darmstadt, Germany; TCI Europe, Zwijndrecht, Belgium). Commercial grade reagents were used without further purification. Reactions were monitored by thin-layer chromatography plates coated with 0.2 mm silica gel 60 F254 (Merck Chemicals, Darmstadt, Germany). TLC plates were visualized by UV irradiation (254 nm). All melting points were determined on a Melting Point B-540 apparatus (Büchi, Flawil, Switzerland) and are uncorrected. IR spectra were recorded on a Nicolet 6700FT-IR spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) over the range of 400–4000 cm−1 using the ATR technique. NMR spectra were measured in CDCl3, DMSO-d6 solution at ambient temperature on a Bruker AvanceTM III 400 spectrometer at frequencies 1H (400 MHz) and 13C (100.26 MHz) or a Bruker AscendTM 500 spectrometer at frequencies 1H (500.13 MHz), 13C (125.76 MHz) (Bruker, Billerica, MA, USA). The chemical shift, δ, was measured relative to CDCl3 7.27 p.p.m. or DMSO-d6 2.5 p.p.m. using tetramethylsilane (TMS) as an internal standard. Coupling constants (J) were all reported in (Hz). Elemental analysis (C, H, N) was performed on an automatic microanalysis Flash 2000 Organic elemental analyzer (Thermo Fisher Scientific, Waltham, MA, USA). Mass spectrometry with high resolution was determined using the “dried droplet” method with a MALDI mass spectrometer LTQ Orbitrap XL (Thermo Fisher Scientific, Waltham, MA, USA) equipped with a nitrogen laser (337 nm, 60 Hz). Spectra were measured in the positive ion mode and in regular mass extent at a resolution of 100,000 at m/z 400. The matrix used was 2,5-dihydrobenzoic acid (DBH).
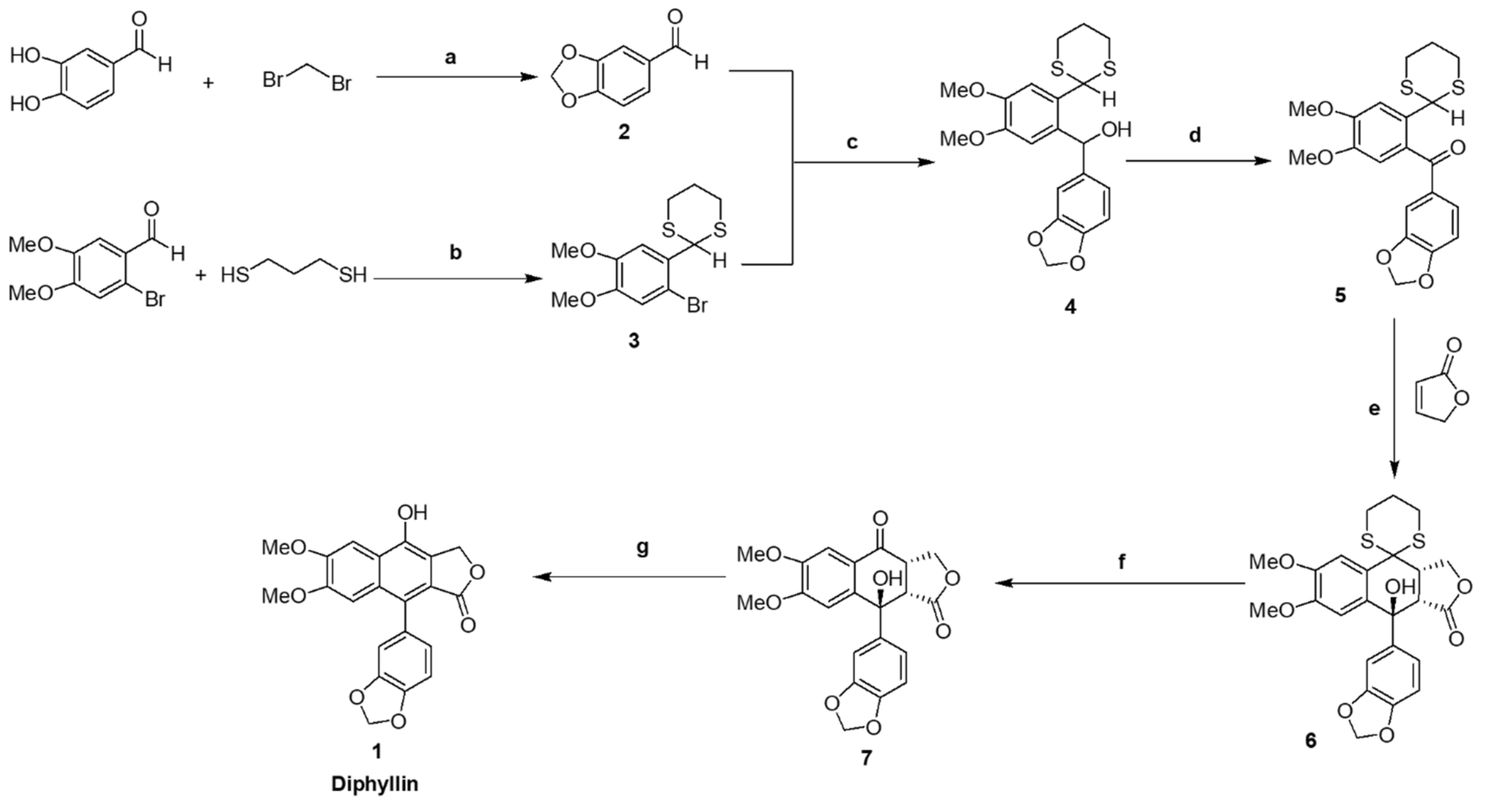
2.2. Synthesis of benzo[d][1,3]dioxole-5-carbaldehyde 2
To a solution of 3,4-dihyroxybenzaldehyde (13.8 g, 99 mmol) in cooled acetonitrile (CH3CN) (200 mL), were added potassium carbonate (K2CO3) (40 g, 289 mmol) and dibromomethane (10,4 mL, 119 mmol). The reaction mixture was then stirred at 90 °C for 24 h. After completion (monitored by TLC), the reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated then extracted with ethyl acetate and water. Thereafter, the organic layer was dried over anhydrous sodium sulfate, concentrated, and the residue purified by column chromatography eluting with ethyl acetate:n-hexane (1:1, v/v) to yield compound 2 (14 g, 94%) as a brown solid. Mp 40–42 °C. IR(ATR): 3081, 3063, 2998, 2985, 2918, 2851, 2793, 2752, 2722, 1669, 1599, 1487, 1446, 1253, 1035, 927, 864, and 811 cm−1. 1H NMR (CDCl3, 500.13 MHz): δ 9.80 (1H, s, -CHO), 7.41 (1H, dd, J = 1.5 Hz, J = 8.0 Hz, Ar-H), 7.33 (1H, d, J = 1.5 Hz, Ar-H), 6.99 (1H, d, J = 8.0 Hz, Ar-H), and 6.07 (2H, s, -O-CH2-O-). 13C NMR (CDCl3, 125.67 MHz): δ 190.4, 153.2, 148.8, 131.9, 128.8, 108.4, 107.0, and 102.2. CHN analysis: Calculated for C8H6O3 (150.13): C, 64.00; H, 4.03. Found: C, 64.07 ± 0.01; H, 4.10 ± 0.02. HRMS: m/z calculated for C8H6O3: 151.03897 [M+H]+; found: 151.03925 [M+H]+.
2.3. Synthesis of 2-(2-bromo-4,5-dimethoxyphenyl-[1,3]dithiane 3
To a round bottom flask containing dry benzene (300 mL) were added 2-bromo-4,5-dimethoxybenzaldehyde (12.3 g, 50.2 mmol), 1,3-propanedithiol (5.04 mL, 50.2 mmol), and p-toluenesulfonic acid (p-TsOH) (0.48 g, 2.79 mmol). The flask was then connected to a Dean-Stark trap and the reaction mixture heated under reflux for 10 h. After completion (monitored by TLC), the reaction mixture was cooled to room temperature, solvent was removed under reduced pressure, and the residue was partitioned between diethyl ether (100 mL), 1M NaOH (100 mL), water (200 mL), and brine (200 mL). The organic layer was subsequently dried over anhydrous sodium sulfate, concentrated, and the residue was purified by column chromatography eluting with ethyl acetate:n-hexane (1:3, v/v) to yield compound 3 (16.7 g, 100%) as a white solid. Mp 125–126 °C. IR (ATR): 3068, 2996, 2965, 2899, 1598, 1505, 1484, 1461, 1449, 1379, 1259, 1250, 1 028, 968, 906, 906, 808, 779, 760, and 578 cm−1. 1H NMR (CDCl3, 400 MHz): δ. 7.15 (1H, s, Ar-H), 6.97 (1H, s, Ar-H), 5.52 (1H, s, -CH2-S-CH-Ar), 3.89 (3H, s, -OCH3), 3,84 (3H, s, -OCH3), 3.12 (2H, brd, J = 12.4 Hz, -S-CH2-CH2-CH2-S-), 2,90 (2H, brdt J = 14.8, 2.8 Hz, -S-CH2-CH2-CH2-S-), 2.17 (1H, m, -S-CH2-CHH-CH2-S-), and 1.93 (1H, m, -S-CH2-CHH-CH2-S-). 13C NMR (CDCl3, 100.26 MHz): δ 149.4, 149.0, 130.2, 115.3, 113.0, 111.1, 56.29, 56.28, 49.3, 32.4, and 25.1. CHN analysis: Calculated for C12H15BrO2S2 (335.28): C, 42.99; H, 4.51; S, 19.13. Found: C, 43.37 ± 0.28; H, 4.45 ± 0.02; S, 18.63 ± 0.14. HRMS: m/z calculated for C12H15BrO2S2: 334.97696 [M+H]+; found: 334.97817 [M+H]+.
2.4. Synthesis of benzo[1,3]dioxol-5-yl-(2-[1,3]dithian-2-yl-4,5-dimethoxyphenyl)-methanol 4
Dithiane 3 (14 g, 41.7 mmol) was dissolved in dry tetrahydrofuran (THF) (200 mL), then n-butyl lithium (n-BuLi) (39 mL, 1.6 mL solution in n-hexane, 62.6 mmol) was added at −78 °C under a N2 atmosphere, after which the reaction mixture was stirred for 1 h. A solution of piperonal 2 (6.89 g, 45.9 mmol) in dry THF (30 mL) was further added at −78 °C, then the reaction mixture was stirred for 2 h, before warming to room temperature over 3 h. After completion (monitored by TLC), the reaction mixture was quenched with a saturated solution of ammonium chloride (90 mL), then extracted with ethyl acetate and water. Thereafter the organic layer was further extracted with brine solution (150 mL), dried over anhydrous sodium sulfate, and evaporated under reduced pressure, giving a crude product residue that was purified by flash chromatography eluting with ethyl acetate:n-hexane (1:3, v/v) to yield compound 4 (15 g, 89%) as a white solid. Mp 75–76 °C. IR (ATR): 3491, 3076, 2996, 2933, 2894, 2848, 2776, 1606, 1509, 1486, 1446, 1440, 1235, 1167, 1082, 1034, 925, 894, 821, 755, 667 cm−1. 1H NMR (CDCl3, 400 MHz): δ. 7.12 (1H, s, Ar-H), 6.88 (1H, d, J = 1.6 Hz, Ar-H), 6.85 (1H, brd, J = 8.0, 1.2 Hz, Ar-H), 6.82 (1H, s, Ar-H), 6.78 (1H, d, J = 8 Hz, Ar-H), 6.14 (1H, s, -Ar-CH-OH-Ar), 5.94 (2H, s, -O-CH2-O-), 5.39 (1H, s, -CH2-S-CH-Ar), 3.90 (3H, s, -OCH3), 3.79 (3H, s, -OCH3), 2.97 (2H, brtd, J = 9.6, 6 Hz, -S-CH2-CH2-CH2-S-), 2.85 (2H, brdt, J = 14.4, 3.6 Hz, -S-CH2-CH2-CH2-S-), 2.58 (1H, s, -Ar-CH-OH-Ar), 2.13 (1H, m, -S-CH2-CHH-CH2-S-), 1.89 (1H, m, -S-CH2-CHH-CH2-S-). 13C NMR (CDCl3, 100.26 MHz): δ 148.96, 148.93, 147.8, 146.9, 137.2, 133.4, 129.0, 120.0, 111.5, 110.6, 108.1, 107.4, 101.1, 72.1, 56.6, 56.0, 48.0, 32.67, 32.60, 25.1. CHN analysis: Calculated for C20H22O5S2 (406.52): C, 59.09; H, 5.45; S, 15.78. Found: C, 58.79 ± 0.15; H, 5.32 ± 0.01; S, 15.63 ± 0.08. HRMS: m/z calculated for C20H22O5S2: 407.09814 [M+H]+; 429.08009 [M+Na]+; found: 407.09865 [M+H]+; 429.08237 [M+Na]+.
2.5. Synthesis of benzo[1,3]dioxol-5-yl-(2-[1,3]dithian-2-yl-4,5-dimethoxyphenyl)-methanone 5
Dimethoxyphenyl alcohol 4 (13 g, 32 mmol) was dissolved in dichloromethane (CH2Cl2) (200 mL), then activated manganese dioxide (MnO2) (48 g, 552 mmol) added. Thereafter the reaction mixture was stirred at room temperature under a N2 atmosphere for 16 h. After completion (monitored by TLC), the reaction mixture was filtered through a plug of Celite and washed with dichloromethane (400 mL). Thereafter, the filtrate was evaporated under reduced pressure to dryness giving compound 5 (12 g, 93%) as a white solid. Mp 150–152 °C. IR (ATR): 3065, 3004, 2918, 2901, 2848, 2828, 1656, 1600, 1513, 1483, 1464, 1436, 1237, 1178, 1033, 955, 892, 827, 765, and 582 cm−1. 1H NMR (CDCl3, 400 MHz): δ 7.35 (1H, d, J = 0.4 Hz, Ar-H), 7.31 (1H, dd, J = 8.0, 2.0 Hz, Ar-H), 7,26 (1H, s, Ar-H), 6.79 (1H, d, J = 8.0 Hz, Ar-H), 6.75 (1H, s, Ar-H), 6.02 (2H, s, -O-CH2-O-), 5.39 (1H, s, -CH2-S-CH-Ar), 3.94 (3H, s, -OCH3), 3.77 (3H, s, -OCH3), 2.89 (2H, brtd, J = 12.4, 2.4 Hz, -S-CH2-CH2-CH2-S-), 2.77 (2H, brdt, J = 14.4, 3.6 Hz, -S-CH2-CH2-CH2-S-), 2.05 (1H, m, -S-CH2-CHH-CH2-S-), and 1.84 (1H, m, -S-CH2-CHH-CH2-S-). 13C NMR (CDCl3, 100.26 MHz): δ 195.0, 152.1, 151.1, 148.1, 147.1, 132.7, 131.6, 129.8, 127.6, 112.1, 111.9, 109.6, 107.8, 102.0, 56.29, 56.22, 47.6, 32.3, and 25.1. CHN analysis: Calculated for C20H20O5S2 (404.50): C, 59.39; H, 4.98; S, 15.85. Found: C, 60.01 ± 0.10; H, 4.99 ± 0.02; S, 15.67 ± 0.05. HRMS: m/z calculated for C20H20O5S2: 405.08249 [M+H]+; 427.06444 [M+Na]+; found: 405.08321 [M+H]+; 427.06552 [M+Na]+.
2.6. Synthesis of 9-benzo[1,3]dioxol-5-yl-9-hydroxy-4-([1,3]dithian-2-yl)-6,7-dimethoxy-3a,4, 4,9a-tetrahydro-3H-naphtho[2,3-c]furan-1-one 6
Benzophenone intermediate 5 (12 g, 29.7 mmol) was dissolved in dry THF (200 mL), lithium hexamethyldisilazide (LiHMDS) (1.0 M in THF, 39 mL) added at −65 °C under a N2 atmosphere, and then the reaction mixture was stirred at −65 °C for 4 h to generate a deep purple anion. Following this, a solution of 2(5H)-furanone (3.2 g, 38.6 mmol, in dry THF) was then added drop by drop over 15 min. Thereafter, the reaction mixture was stirred at −65 °C under a N2 atmosphere for another 48 h. After completion (monitored by TLC), the reaction mixture was warmed to room temperature for 1 h and quenched with H2O (5 mL), before the organic solvent was evaporated under reduced pressure. The resulting crude product residue was then purified by column chromatography eluting with ethyl acetate:n-hexane (4:1, v/v) to yield compound 6 (3.7 g, 26%) as a white solid. Mp 205–207 °C. IR (ATR): 3503, 3087, 2973, 2935, 2904, 2847, 2825, 1766, 1741, 1608, 1514, 1502, 1481, 1464, 1454, 1435, 1372, 1361, 1301, 1263, 1235, 1173, 1081, 1062, 1018, 973, 948, 893, 818, 785, 729, and 636 cm−1. 1H NMR (DMSO-d6, 500.13 MHz): δ 7.43 (1H, s, Ar-H), 6.86 (1H, d, J = 8.0 Hz, Ar-H), 6.80 (1H, d, J = 1.5 Hz, Ar-H), 6.74 (1H, dd, J = 7.0, 2.0 Hz, Ar-H), 6.40 (1H, s, Ar-H), 6.08 (1H, s, -Ar-C-OH-CH-), 6.00 (2H, s, -O-CH2-O-), 4.30 (1H, m, -C-CH-CH2-O-), 4.06 (2H, m, -C-CH-CH2-O-), 3.77 (3H, s, -OCH3), 3.51 (3H, s, -OCH3), 3.43 (1H, brtd, J = 13.7, 2.5 Hz, -S-CH2-CH2-CHH-S-), 3.27 (1H, brt, J = 13.5 Hz, -S-CH2-CH2-CHH-S-), 3.08 (1H, brd, J = 6.0 Hz, -Ar-C-OH-CH-CO), 2.87 (1H, brd, J = 15.0 Hz, -S-CHH-CH2-CH2-S-), 2.71 (1H, brd, J = 14.5 Hz, -S-CHH-CH2-CH2-S-), 2.17 (1H, brd, J = 14.5 Hz, -S-CH2-CHH-CH2-S-), and 1.77 (1H, brq, J = 13.0 Hz, -S-CH2-CHH-CH2-S-). 13C NMR (DMSO-d6, 125.76 MHz): δ 174.2, 148.7, 148.5, 146.7, 145.7, 143.6, 133.8, 126.4, 119.3, 112.5, 109.4, 107.5, 106.7, 101.0, 69.9, 68.3, 55.5, 55.3, 51.0, 49.0, 41.0, 28.1, 26.2, and 23.7. CHN analysis: Calculated for C24H24O7S2 (488.57): C, 59.00; H, 4.95; S, 13.13. Found: C, 58.68 ± 0.18; H, 5.23 ± 0.02; S, 12.82 ± 0.07. HRMS: m/z calculated for C24H24O7S2: 511.08557 [M+Na]+; 527.05950 [M+K]+; found: 511.08816 [M+Na]+; 527.06223 [M+K]+.
2.7. Synthesis of 9-benzo[1,3]dioxol-5-yl-9-hydroxy-6,7-dimethoxy-3,3a,9,9a-tetrahydronaph- -tho[2,3-c]furan-1,4-dione 7
A solution of lactone 6 (3.5 g, 7.1 mmol) in aqueous acetonitrile (85%, v/v, 50 mL) with mercuric oxide (HgO) (1.6 g, 7.8 mmol) and mercury chloride (HgCl2) (4.2 g, 15.7 mmol) was heated under reflux for 5 h. After completion (monitored by TLC), the reaction mixture was cooled to room temperature, filtered through a Celite bed, and the filtrate evaporated under reduced pressure. The resulting crude product residue was then dissolved in chloroform (150 mL), and extracted with a saturated solution of ammonium carbonate. Thereafter the organic layer was extracted with brine solution (150 mL), dried over anhydrous sodium sulfate and evaporated under reduce pressure. The resulting crude product residue was purified by flash chromatography eluting with ethyl acetate:n-hexane (2:3, v/v) to yield compound 7 (1.257 g, 44.2%) as a white solid. Mp 103–104 °C. IR (ATR): 3451, 2966, 2919, 2850, 2640, 1747, 1735, 1665, 1593, 1505, 1484, 1460, 1436, 1364, 1320, 1278, 1239, 1212, 1145, 1129, 1096, 1080, 1031, 1010, 987, 904, 820, 794, 765, 723 cm−1. 1H NMR (CDCl3, 400 MHz): δ 7.48 (1H, s, Ar-H), 7.25 (1H, s, Ar-H), 6.86 (1H, d, J = 2.0 Hz, Ar-H), 6.65 (1H, d, J = 8.0 Hz, Ar-H), 6.41 (1H, dd, J = 8.4, 2.5 Hz, Ar-H), 5.94 (2H, s, -O-CH2-O-), 5.74 (1H, s, -Ar-C-OH-CH-), 4.73 (1H, d, J = 9.6 Hz,-CO-CH-CHH-O-), 4.30 (1H, dd, J = 9.2, 5.6 Hz, -CO-CH-CHH-O-), 3.96 (3H, s, -OCH3), 3.94 (3H, s, -OCH3), 3.44 (1H, d, J = 7.2 Hz, -Ar-C(OH)-CH-CO-O-), and 3.09 (1H, dd, J = 5.6, 4.8 Hz,-CO-CH-CH2-O-). 13C NMR (CDCl3, 100.26 MHz): δ 193.0, 176.9, 155.9, 149.7, 148.2, 147.6, 141.2, 138.5, 125.5, 120.1, 108.8, 108.1, 107.9, 107.0, 101.5, 72.5, 70.8, 56.5, 56.2, 50.4, and 46.1. CHN analysis: Calculated for C21H18O8 (398.36): C, 63.32; H, 4.55. Found: C, 63.21 ± 0.03; H, 4.69 ± 0.01. HRMS: m/z calculated for C21H18O8: 421.08939 [M+Na]+; 437.06333 [M+K]+; found: 421.09018 [M+Na]+; 437.06448 [M+K]+.
2.8. Synthesis of 9-(3‣,4‣-methylenedioxyphenyl)-4-hydroxy-6,7-dimethoxynaphtho[2,3-c]furan-1(3H)-one (Diphyllin) 1
Benzofuranone
7 (1.084 g, 2.7 mmol) and
p-TsOH (0.362 g, 1.9 mmol) were heated under reflux in benzene (80 mL) for 15 h. After completion (monitored by TLC), the reaction mixture was cooled to room temperature and the solvent removed under reduced pressure. The crude product residue was then purified by flash chromatography eluting with ethyl acetate:
n-hexane (1:1,
v/v) to yield compound
1 (0.730 g, 70%) as a yellow solid that was recrystallized. Mp 275–277 °C. IR (ATR): 3197, 3006, 2955, 2923, 2853, 2648, 1703, 1613, 1594, 1507, 1492, 1455, 1433, 1358, 1332, 1227, 1211, 1192, 1169, 1125, 1084, 1035, 1007, 930, 948, 915, 860, 768, 730, and 719 cm
−1.
1H NMR (DMSO-d
6, 400 MHz): δ. 10.4 (1H, s, Ar-O
H), 7.61 (1H, s, Ar-
H), 7.01 (1H, d,
J = 8.0 Hz, Ar-
H), 6.95 (1H, s, Ar-
H), 6.86 (1H, d,
J = 1.6 Hz, Ar-
H), 6.75 (1H, dd,
J = 10.0, 5.6 Hz, Ar-
H), 6.11 (2H, s, -O-C
H2-O), 5.35 (2H, s, -O-C
H2-Ar), 3.94 (3H. s, -OC
H3), 3.65 (3H, s, -OC
H3).
13C NMR (DMSO-d
6, 100.26 MHz): δ 169.6, 150.4, 149.6, 146.8, 146.6, 144.8, 129.5, 129.4, 128.8, 123.7, 123.2, 121.6, 118.6, 111.0, 107.8, 105.4, 101.0, 100.7, 66.6, 55.5, and 55.1. CHN analysis: Calculated for C
21H
16O
7 (380.35): C, 66.31; H, 4.24. Found: C, 66.45 ± 0.10; H, 4.56 ± 0.02. HRMS:
m/z calculated for C
21H
16O
7: 381.09688 [M+H]
+; 403.07882 [M+Na]
+; 419.05276 [M+K]
+; found: 381.09809 [M+H]
+; 403.08028 [M+Na]
+; 419.05421 [M+K]
+. Previously reported data [M−H]
+ 379.0825 [
24].
2.9. Viruses, Cells, and Compounds
Our antiviral studies were performed using typical representatives of the following virus families: (i)
Flaviviridae (TBEV, strain Hypr, a representative of the European TBEV subtype, provided by the Collection of Arboviruses, Institute of Parasitology, Biology Centre of the Czech Academy of Sciences, Ceské Budějovice, Czech Republic (
http://www.arboviruscollection.cz/index.php?lang=en, accessed on 10 January 2022); ZIKV, the Brasilian strain Paraiba_01; kindly provided by Prof. Paolo M. de A. Zanotto, University of São Paulo, Brasil; WNV, strain Eg-101, provided by the Collection of Arboviruses, Institute of Parasitology, Biology Centre of the Czech Academy of Sciences, Ceske Budejovice, Czech Republic); (ii)
Phenuiviridae (RFVF, strain H13/96, kindly provided by Karel Bilek, National Institute of Nuclear, Chemical and Biological Protection, Czech Republic); (iii)
Rhabdoviridae (RABV, strain CVS-11, kindly provided by Prof. Anthony R. Fooks, Animal and Plant Health Agency, UK); and (iv)
Herpesviridae (HSV-1, strain MacIntyre, kindly provided by Prof. Andreas Sauerbrei, German Reference Laboratory of HSV und VZV, Germany).
Vero cells (ATCC CCL-81, African Green Monkey, adult kidney, epithelial), baby hamster kidney cells (BHK-21, ATTC CCL-10), and human hepatocarcinoma cells (Huh-7) were grown in Dulbecco′s modified Eagle′s medium (DMEM); porcine kidney stable (PS) [
27] were cultured in Leibovitz (L-15) medium; human brain cortical astrocytes (HBCA, ScienCell, Carlsbad, CA, USA) were cultivated in Astrocyte medium; human neuroblastoma UKF-NB-4 cells [
28] were cultured in Iscove’s modified Dulbecco’s medium (IMDM). The media were supplemented with 3% (L-15), 6% (Astrocyte medium), or 10% (IMDM and DMEM) newborn calf serum plus 100 U/mL penicillin, 100 µg/mL streptomycin, and 1% glutamine (Sigma-Aldrich, Prague, Czech Republic). Vero, HBCA, UKV-NB-4, Huh-7, and BHK-21 cells were cultured at 37 °C under 5% CO
2, whereas PS cells were cultivated at 37 °C under a normal atmosphere (without CO
2 supplementation).
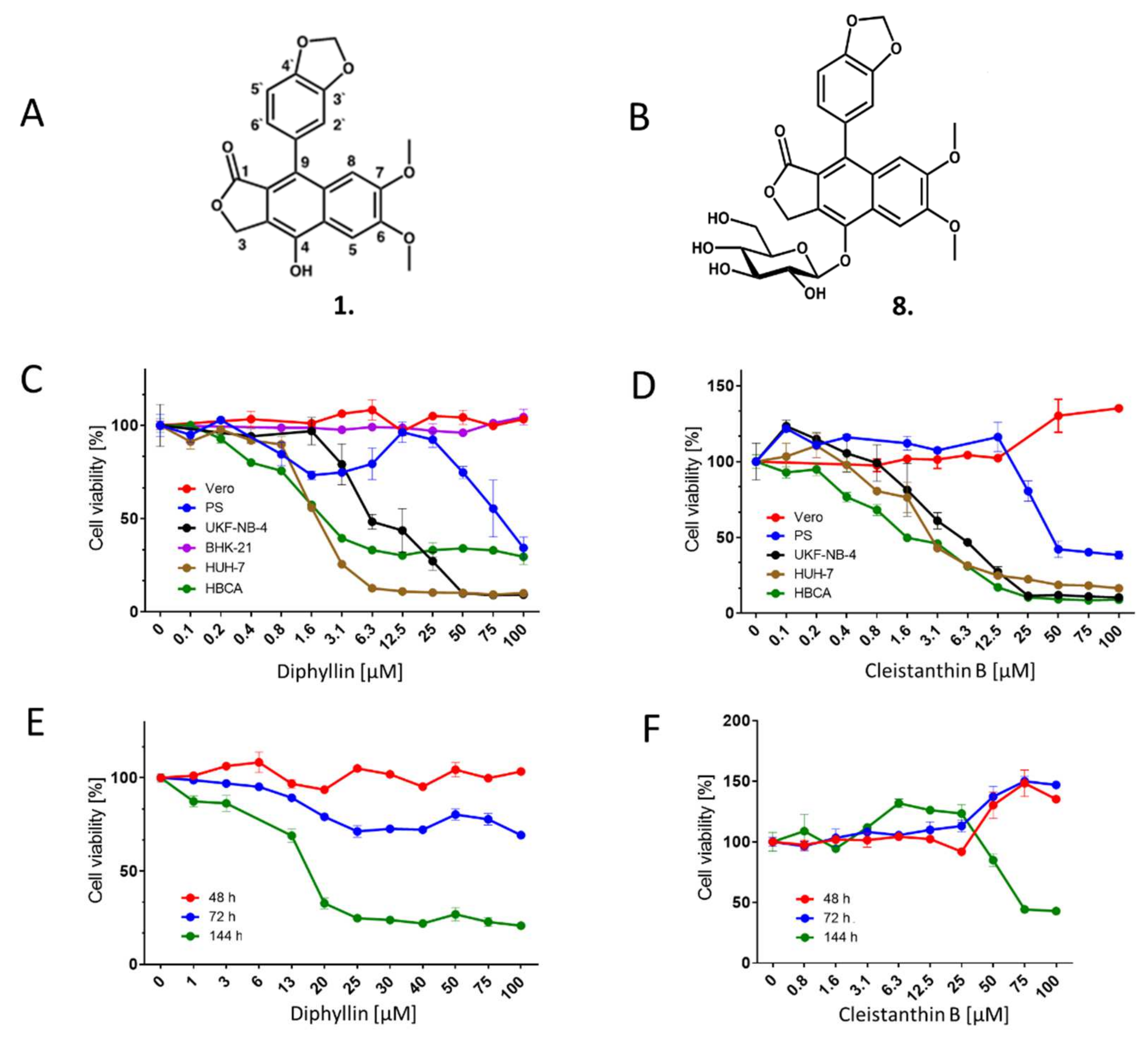
For all antiviral/cytotoxicity assays, we used in-house synthesized diphyllin
1 (
Scheme 1,
Figure 1A), whose biological activities were compared with the commercially synthesized compound purchased from Apigenex (Prague, Czech Republic) (
Figure S1). To compare the biological activities of both compounds, we performed a viral titer reduction assay; virus and compound were added simultaneously to Vero cells and cultivated for 48 h at 37 °C under 5% CO
2 (see
Section 2.11. simultaneous treatment). The diphyllinoside known as diphyllin-4-β-D-glucopyranoside (cleistanthin B)
8 was obtained from WuXi AppTec (Tianjin, China). All compounds were solubilized in dimethyl sulfoxide (DMSO) as 10 mM stock solutions.
2.10. Cytotoxicity Assays
To determine the cytotoxicities of diphyllin 1 and diphyllinoside cleistanthin B 8, we used multiple cells/cell lines of neural and extraneural origin. Vero, PS, UKF-NB-4, Huh-7, or HBCA cells were seeded in 96-well microtitration plates (2 × 104 cells/well), and incubated for 24 h at 37 °C. After incubation, diphyllin 1 or cleistanthin B 8 were added to the cells (0 to 100 µM) then treated cells were further cultivated for 48 h at 37 °C. Cytotoxicity of diphyllin 1 for BHK-21 cells was measured after 72 h cultivation. In order to evaluate Vero cell cytotoxicities from exposure to diphyllin 1, the cells were seeded in 96-well microtitration plates (2 × 104 cells/well), incubated for 24 h at 37 °C under 5% CO2, and then diphyllin 1 or cleistanthin B 8 (0 to 100 µM) was added. The treated cells were further cultivated for 48, 72, and 144 h at 37 °C under 5% CO2. Cytotoxicity of bafilomycin A1 (0 to 200 µM) was assayed in Vero cells after a 48-h incubation at 37 °C under 5% CO2. The cytotoxicity measured in terms of cell viability was determined with Cell Counting Kit-8 (Dojindo Molecular Technologies, Munich, Germany), according to the manufacturer’s instructions. The respective concentrations of each compound under investigation that reduced cell viability by 50% (CC50 values) were determined.
2.11. Antiviral Assays
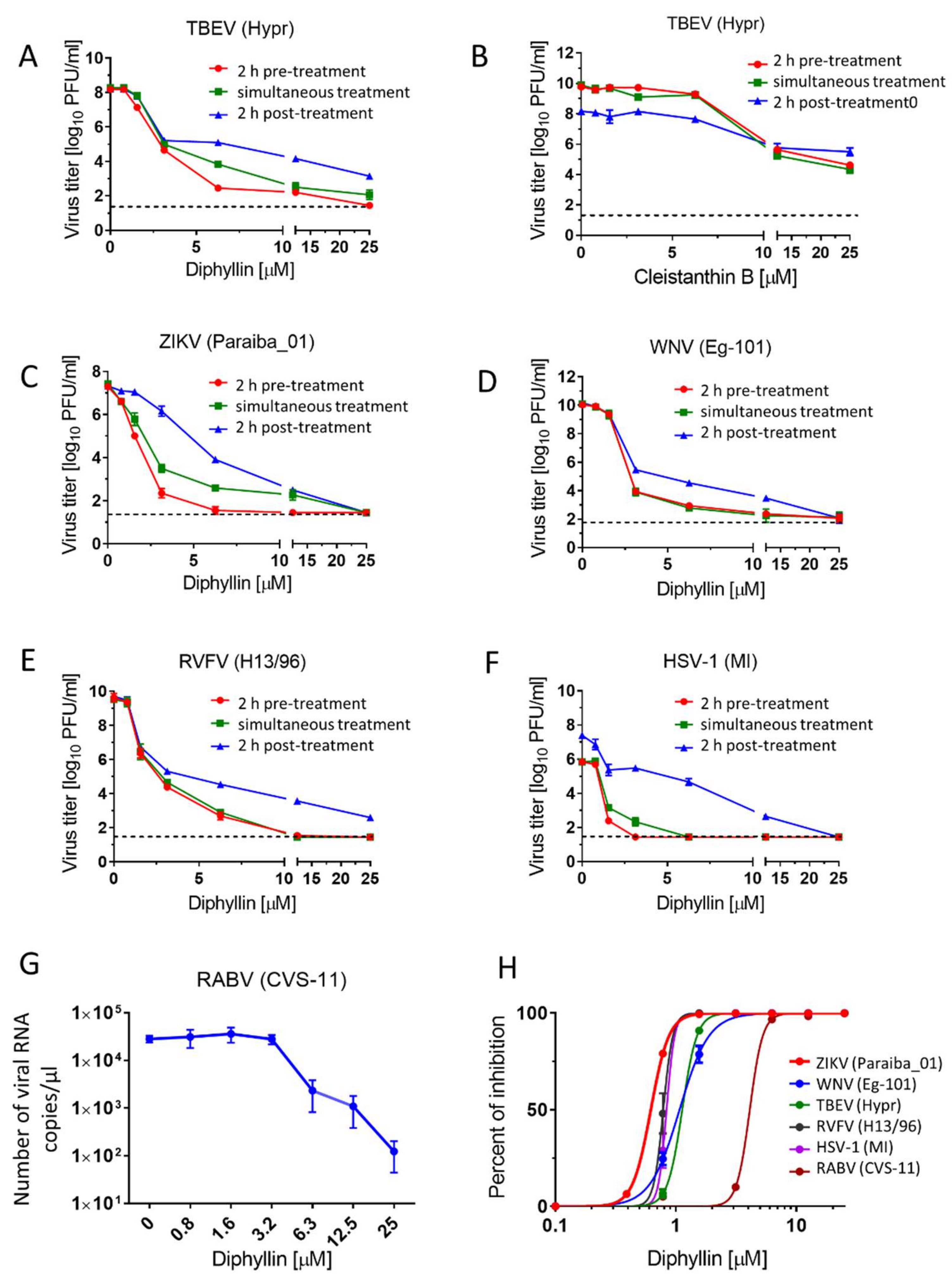
Viral titre reduction assays were performed to determine the sensitivity of the tested viruses to diphyllin 1 in cell culture. Vero cells were used for the evaluation of the antiviral efficacy of diphyllin 1 against TBEV, WNV, ZIKV, RVFV, and HSV-1. Host cells were seeded in 96-well plates (approximately 2 × 104 cells per well) and incubated for 24 h at 37 °C under 5% CO2 to form a confluent monolayer.
For each virus, two independent experiments were performed in triplicate in each case. In each independent experiment, infected Vero cells were treated with drug at three different times, as follows: (i) 2 h pre-treatment assays—media (200 µL) with diphyllin 1 (0 to 25 µM) (2-fold dilution, three wells per concentration) were added to cell monolayers 2 h prior to infection. After 2 h incubation at 37 °C under 5% CO2, media were aspirated and replaced with fresh compound-containing media (200 µL) in the same concentration range, then inoculated with the appropriate virus at an MOI of 0.1. Cells were further incubated for 48 h at 37 °C under 5% CO2; (ii) simultaneous treatment assays—media containing diphyllin 1 (0 to 25 µM) were inoculated with the appropriate virus (MOI of 0.1) and added to cells that were then incubated for 48 h at 37 °C under 5% CO2; (iii) 2 h post-treatment assays—Vero cells were first infected with the appropriate virus (MOI of 0.1), and 2 h after infection (the time needed for virus adsorption and internalization), media containing diphyllin 1 (0 to 25 µM) were added to infected cells that were then incubated for 48 h at 37 °C under 5% CO2.
Following incubation, media were collected and viral titers were determined by plaque assay (see below) to construct dose-dependent inhibition curves. Since the greatest inhibitory effects were observed in 2 h pre-treatment assays, then titer values determined from these assays were also used to estimate 50% effective concentration (EC50) values. The antiviral activity of cleistanthin B 8 was determined only for TBEV in Vero cells, using the same protocol as described above. Media were collected 48 h after infection, and TBEV titers were estimated using plaque assays once again.
For anti-RABV assays, a suspension of BHK-21 cells (5 × 104 cells per a well) was incubated with diphyllin 1 (0 to 25 µM) and with RABV (106.58 TCID50/mL) at 37 °C under 5% CO2 for 40 min, mixing every 10 min. Following incubation, the RABV-infected cells were seeded on 96 well plates and cultivated for 72 h at 37 °C under 5% CO2 (a longer incubation time was used compared with other viruses tested since RABV does not typically generate sufficiently high titers when incubated for 48 h). Thereafter, media samples were collected, and the RABV RNA was quantified using quantitative real-time PCR (RT-qPCR). Two independent experiments were performed in triplicate (as above).
Antiviral assays with bafilomycin A1 were performed with Vero cells seeded in 96-well plates (2 × 104 cells per well) and incubated for 24 h at 37 °C under 5% CO2 to form a confluent monolayer. Media containing bafilomycin A1 (0 to 100 nM) were simultaneously infected with ZIKV (strain Paraiba_01) or HSV-1 (strain MacIntyre) (MOI of 0.1) and added to cells that were then incubated for 48 h at 37 °C under 5% CO2. Viral titers were determined by plaque assays.
2.12. Plaque Assay
To quantifiy the viral titres for ZIKV, WNV, RVFV, and HSV-1, plaque assays were performed using Vero cells. For TBEV viral titer determination, we performed PS cell-based plaque assays. In both cases, a modified protocol was used as originally developed by De Madrid and Porterfield [
29]. Briefly, 10-fold dilutions of the virus were prepared in 24-well tissue culture plates, and the cells were added to each well (0.6–1.5 × 10
5 cells/well). After 4 h incubation, the suspension was overlaid with 3% (
w/v) carboxymethylcellulose in DMEM (for Vero cells) or L15 (for PS cells). Following day 5 of incubation at 37 °C and 5% CO
2 (DMEM) or in a normal atmosphere (L15), the infected plates were washed with phosphate-buffered saline, and the cell monolayers were stained with naphthalene black. The virus titer was expressed as plaque-forming units (PFU)/mL.
2.13. Quantitative Reverse Transcription PCR (RT-qPCR)
RABV RNA was isolated from growth media supernatants using the QIAmpViral RNA mini kit (Qiagen, Germantown, MD, USA) following the manufacturer’s instructions. RT-qPCR measurements were performed on the LightCycler 480 II in a 96-well plate block (Roche, Basel, Switzerland) using the Advanced kit for Tick-borne encephalitis (Genesig, Germantown, MD, USA) and Lyophilised OneStep qRT-PCR (Oasig) according to manufacturer’s instructions. RABV RNA copy numbers/mL was calculated from calibration curves based on standards provided by the manufacturer (Genesig, Germantown, MD, USA).
2.14. Immunofluorescence Staining
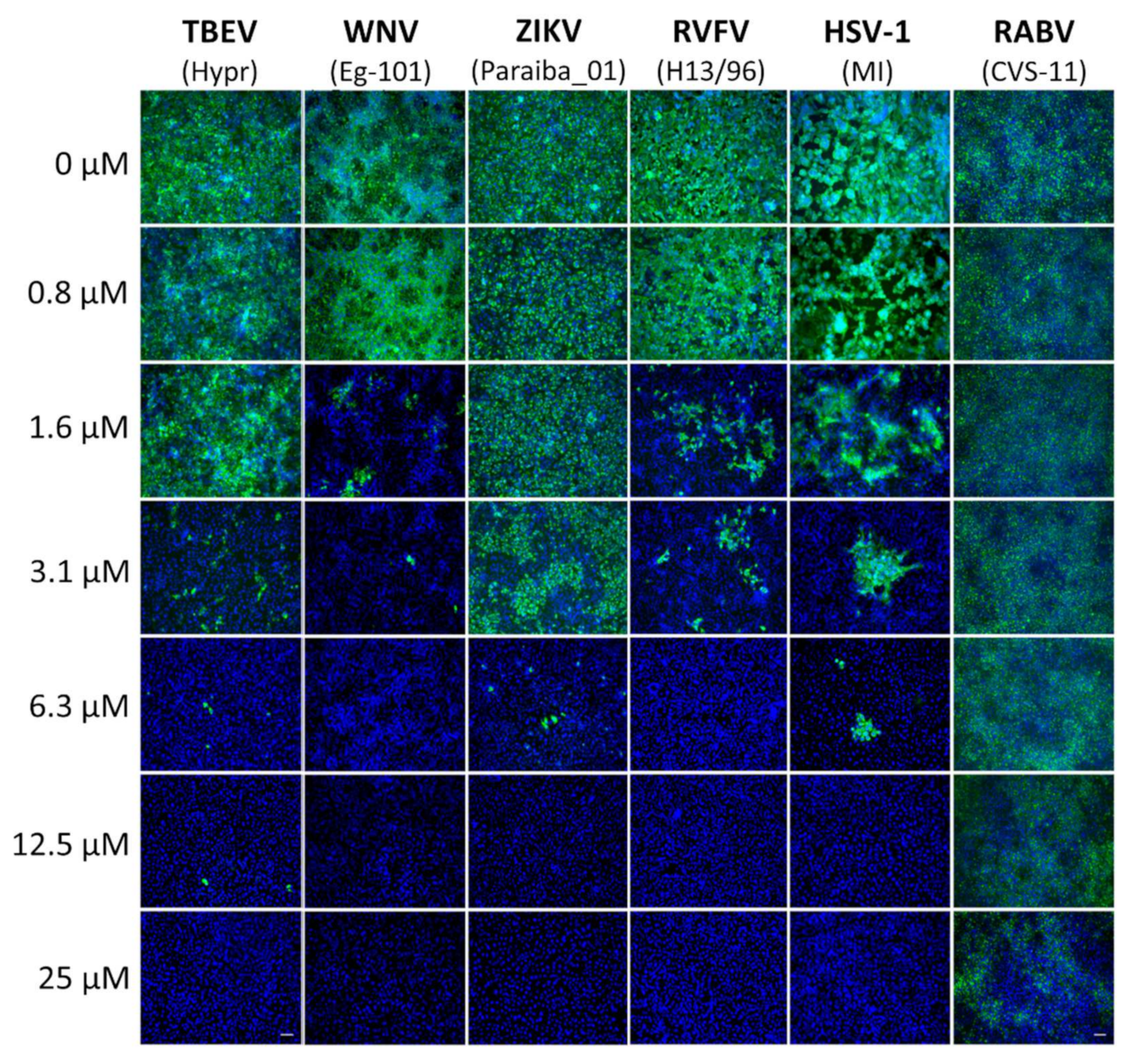
In order to measure the compound-induced inhibition of viral antigen expression, a cell-based immunostaining assay for TBEV, WNV, ZIKV, RVFV, RABV, and HSV-1 was performed. Vero cells were used for the cultivation of TBEV, WNV, ZIKV, RVFV, and HSV-1 and BHK-1 cells for RABV cultivation.
Vero cells were seeded onto 96-well microtitration plates, cultivated for 24 h, and then treated with diphyllin 1 (0 to 25 μM) (2-fold dilution, three wells per concentration) for 2 h. Thereafter, cell monolayers were infected with the appropriate virus (MOI of 0.1) and cultured for 48 h at 37 °C under 5% CO2. After incubation, the cells were fixed with cold acetone:methanol (1:1, v/v) and blocked with 10% fetal bovine serum. For flavivirus immunostaining (i.e., TBEV, WNV, and ZIKV), cells were incubated with a mouse monoclonal antibody targeting the flavivirus group surface antigen (protein E) (1:250; antibody clone D1-4G2-4-15; Sigma-Aldrich, Prague, Czech Republic). Anti-Herpes Simplex Virus Type 1/2 antibody (1:250; antibody clone M4091313) and Anti-Nucleoprotein antibody (1:250; AA 1-245, UniProt: P21700) were obtained from Antibodies-online GmbH (Aachen, Germany) and were used for the immunostaining of HSV-1 and RVFV, respectively. Subsequently, cells were labeled with an anti-mouse goat secondary antibody conjugated with fluorescein isothiocyanate (FITC; 1:500; Sigma-Aldrich, Prague, Czech Republic) following 1 h incubation at 37 °C. Cells were then counterstained with 4′,6-diamidino-2-phenylindole (DAPI; 1 μg/mL; Sigma-Aldrich, Prague, Czech Republic) to visualize cell nuclei. Fluorescence signals were recorded using an Olympus IX71 epifluorescence microscope (Olympus Corporation, Tokyo, Japan).
For the immunostaining of RABV-infected cells, a suspension of BHK-21 cells (5 × 104 cells per well) was incubated with diphyllin 1 (0 to 25 µM) (2-fold dilution, three wells per concentration) and RABV (106.58 TCID50/mL) at 37 °C for 40 min, mixing every 10 min. Following incubation, the RABV-infected cells were seeded on a 96 well plate and cultivated for 72 h. After fixation with cold acetone–methanol (1:1, v/v) and blocking with 10% fetal bovine serum, the cells were incubated with an Anti-Rabies-Specific monoclonal antibody (1:250; Art.No. PA1202; Enzo Life Science, NY, USA) labeled with FITC (green). As above, cell nuclei were counterstained with DAPI (1 μg/mL), and fluorescence signals were recorded, also as described above.
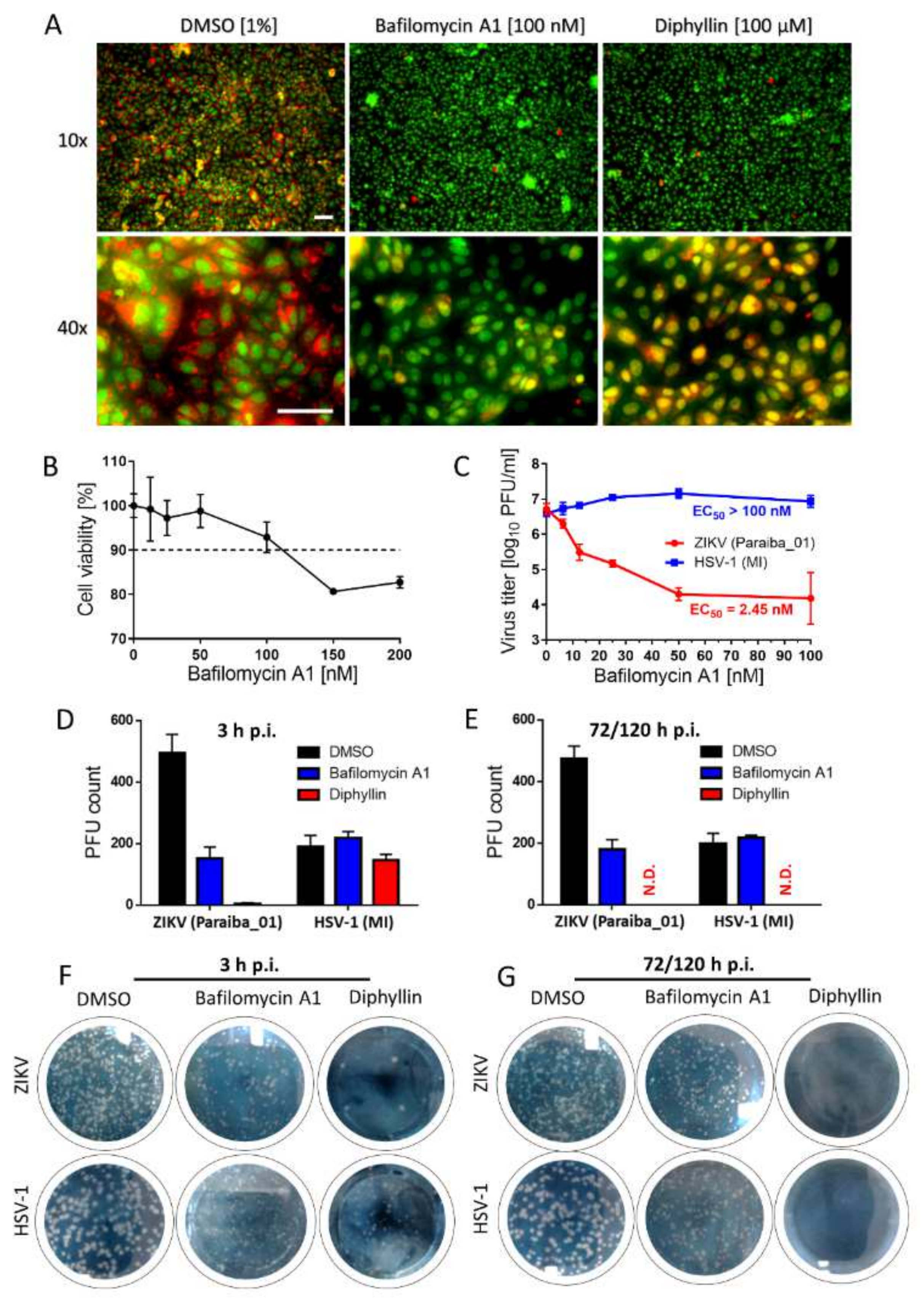
2.15. Analysis of Endosomal Acidification Effects
Vero cells (1 × 106 cells/well) were cultured in a 6-well microtiter plate for 24 h. Then, the growth medium was aspirated and replaced with a fresh medium containing diphyllin 1 (100 µM) or bafilomycin A1 (100 nM) at 37 °C for 20 min. Bafilomycin A1 was used as a positive control, and DMSO 1% (v/v) was applied as a negative control. Acridine orange dye (1 μg/mL; Sigma-Aldrich) was added to each well and incubated at 37 °C for 10 min. Fluorescence signals were recorded using an Olympus IX71 epifluorescence microscope (Olympus Corporation, Tokyo, Japan). The red and green fluorescence signals were acquired using Texas Red and FITC excitation filters, respectively.
2.16. Mechanistic Analysis of Diphyllin-Mediated Antiviral Effects
Vero cells were seeded in 6-well plates and incubated for 24 h at 37 °C under 5% CO2 until confluent. Thereafter, a virus inoculum (106 PFU/mL, ZIKV strain Paraiba_01 or HSV-1 strain MacIntyre) and diphyllin 1 (100 µM) or bafilomycin A1 (100 nM) were added to the cells and incubated for 3 h. Following incubation, Vero cells were washed with culture medium to remove the compounds, and the non-adsorbed virus and fresh medium with 4% (w/v) carboxymethylcellulose were added to the cells. After further incubation for 72 (with HSV-1) or 120 h (with ZIKV), Vero cell monolayers were stained with naphthalene black, and the number of plaques was counted. Control cells were not washed, and the compounds and virus were retained in cell culture until the end of the experiment. Following a 3 h incubation, 4% (w/v) carboxymethylcellulose was added to the cells, and the cells were incubated for 72 or 120 h, as described above. Then, the monolayers were stained with naphthalene black, and the number of plaques was counted.
4. Discussion
Since diphyllin
1 is very often found in plants in the form of glycosides, initial efforts were made to cross-compare biological activities of diphyllin
1 with the glycoside analogue cleistanthin B
8. Prior to full antiviral studies, the cytotyoxicity profiles of diphyllin
1 and cleistanthin B
8 were studied in a range of cells lines. Vero and BHK-1 cells were resistant to the administration of both compounds. However, both compounds were cytotoxic for PS, UKF-NB-5, and Huh-7 cell lines, and also for primary human astrocytes HBCA (
Figure 1,
Table 1). Indeed, diphyllin
1 has been found cytotoxic to many other cell lines besides such as Mardin-Darby canine kidney cells (MDCK, CC
50 of 3.48 µM) [
22], human lung carcinoma cells (A549, CC
50 of 24.01 µM) [
22], feline macrophage-like cells (fcwf-4, CC
50 of 5.99 µM) [
17], and rabbit lung cells (RL-33 CC
50, of 63 µg/mL) [
25]. Given the anticancer capabilities of diphyllin
1 and analogs [
7,
8,
32,
33,
34,
35,
36], such activities against multiple immortalized cell lines should not perhaps be so surprising. Accordingly, diphyllin is similar to other V-ATPase blockers, which were recently described to have significant anticancer potencies indicative of general anticancer drugs. These include, plecomacrolide antibiotics, classical V-ATPase inhibitors such as concanamycin and bafilomycin A1, benzolactone enamides (apicularens, lobatamides, oximidines, and cruentaren), archazolid, indolyls, and late-generation V-ATPase inhibitors, such as FR202126, 3-bromopyruvate, or tributyltin chloride [
37]. Importantly, the antiviral effects of diphyllin
1 are observed at considerably lower (non-cytotoxic) concentrations.
Here we demonstrate that diphyllin
1 acts as a sub-micromolar or low-micromolar inhibitor of multiple enveloped RNA viruses from the
Flaviviridae, Phenuiviridae, and
Rhabdoviridae families (
Figure 2,
Table 2), while cleistanthin B
8 displayed significantly lower antiviral effects compared with those for diphyllin
1 and was not therefore studied further here. In comparison, a diphyllin
1 and the diphyllin analog patentiflorin A have been shown previously to block ZIKV replication in vitro and prevented mortality in a rodent in vivo model of ZIKV infection as alluded to above [
13]. In contrast with our study, the ZIKV infectivity in diphyllin
1-treated cells was evaluated using immunofluorescence staining-based method demonstrating the anti-ZIKV effect of diphyllin
1 even in nanomolar concentrations [
13]. Furthermore, analogs patentiflorin A and justiprocumin B, were found to display nanomolar activity against four clinical HIV-1 isolates in human peripheral blood mononuclear cells (PBMC) cells [
23,
24] as well broad spectrum nanomolar inhibition of HIV-1 strains resistant to several reverse transcriptase inhibitors [
24]. In addition, diphyllin
1 itself has been shown to inhibit the replication of feline infectious peritonitis virus (FIPV), a member of the
Coronaviridae family, in fcwf-4 cells. Furthermore, diphyllin
1 has also been shown to be potent in vitro against influenza virus in MDCK cells [
22]. Some other diphyllin derivatives (e.g., justicidin A and B, and some others) have also been found active against vesicular stomatitis virus (VSV) when tested in RL-33 cells [
25]. Therefore, taken together with our data here, there can be no doubt that diphyllin
1 is a potentially useful broad antiviral agent through its broad cellular cytotoxicity profile suggests that this antiviral agent would be best targeted to sites of viral infection by means of drug delivery nanoparticles and other nanomedicine approaches [
17].
In mechanistic terms, the success of diphyllin
1 as a broad antiviral agent, as demonstrated here using complementary in vitro virus infection models, might seem somewhat paradoxical. Given that diphyllin
1 is a known V-ATPase inhibitor and hence inhibitor of endosome acidification, then this mechanism of action should account for the inhibition of the replication cycle of viruses that enter host cells by endocytosis followed by acid pH-induced membrane fusion [
13,
17,
22]. This is certainly true of
Flaviviridae, Bunayviridae, and
Rhabdoviridae families. All these viruses enter host cells through endocytosis and require low pH-induced fusion of viral envelope with endosome membrane to continue their replication cycles [
16]. Consistent with this argument, our data here indicate clearly that the mechanism of action of diphyllin
1 against ZIKV, a member of the Flaviviridae family, takes place during the early phases of the virus replication cycle on a time scale consistent with a mechanism of action involving V-ATPase inhibition and the direct modulation of endosome acidification to abrogate the virus replication cycle, all in keeping with the known mechanism of ZIKV host cell entry by endocytosis [
13].
On the other hand, diphyllin
1 also exhibits sub-micromolar antiviral potency against HSV-1, a virus that enters cells by plasma membrane interactions and cell surface fusion (not via endocytosis) at neutral pH (with no need for V-ATPase activity) [
16]. Hence, our observed diphillin-mediated anti-HSV-1 effects cannot be based on the inhibition of HSV-1 early cell entry but rather on the suppression of other, later stages of the HSV-1 replication cycle. Accordingly, diphillin
1, in marked contrast to bespoke V-ATPase inhibitors such as bafilomycin A1, must be able to employ additional, alternative antiviral mechanisms of action to abrogate viral replication cycles at different stages as appropriate. Indeed, besides being a V-ATPase inhibitor, diphyllin
1 and diphyllinosides have also been shown to target topoisomerase IIα [
38], to induce apoptosis and protective autophagy through reactive oxygen species production [
8], block voltage-gated K
+ channels in mouse neuroblastoma cells [
39], stimulate interferon-γ production [
40], reduce nitric oxide levels [
41], and modulate TNF-α, and IL-12 production in mouse macrophages [
12]. Hence, we speculate that any or even some of these alternative capabilities might be used to suppress viral infection cycles that do not begin with endocytosis followed by acid pH-induced fusion of viral and endosomal membranes.
Besides diphyllin
1 and bafilomycin A1, broad-spectrum antiviral activity was previously demonstrated also for other V-ATPase blockers, such as saliphenylhalamide (SaliPhe), tenatoprazole, esomeprazole, and ammonium chloride, to name a few. The expected molecular target for SaliPhe is a binding site of the proton translocation domain of cellular V-ATPase; this compound can effectively inhibit several wild types of influenza viruses [
42]. A similar mode of action was also considered for ammonium chloride, which showed inhibitory activity against human rhinoviruses [
43]. In addition, ammonium chloride blocked infection of human papillomavirus (PV) type 16 by preventing the transport of PV viral particles from early endosomes to caveosomes [
44]. Multiple modes of antiviral action were also described for tenatoprazole and esomeprazole. In addition to proton pump blocking, these compounds were observed to inhibit the budding of some enveloped viruses (e.g., HIV-1, Ebola virus, or Dengue virus) by blocking the interaction of Tsg101 protein, a member of the host endosomal sorting complex required for transport (ESCRT), with ubiquitin. Inhibition of Tsg101-ESCRT interaction blocks delivery of ESCRT to budding viral sites, causing the suppression of the virus particle release by membrane scission [
45]. It is clear from these examples that different compounds, originally established as classical proton pump inhibitors, can interact with different cellular targets, resulting in a strong affecting on the viral replication process. This phenomenon probably also applies to diphyllin
1.
Finally, we note that in our efforts to uncover the range and extent of the antiviral activities of diphyllin
1, as described here, we also outlined a revised, updated, and improved synthesis of diphyllin
1 to support these in vitro studies. The further benefits of this synthesis are two-fold: (1) it will enable the cost-effective provision of much larger quantities of this compound for future in vivo follow-up experiments using animal models of virus infections; and (2) it will make possible the preparation of new in house diphyllin analogs suitable for future structure-activity correlation experiments. In particular, the synthesis will make possible the relatively facile preparation of new diphyllin analogs through functional group substitutions of at C-3, C-4, C-2′, C-3′, C-4′, C-5′, and C-6′ (see
Figure 1A). We expect to report the results of such structure-activity correlation experiments in due course, building on the results of the studies described here.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}