
Rapid SARS-CoV-2 Intra-Host and Within-Household Emergence of Novel Haplotypes

 , ,
, ,  ,
,  , , , ,
, , , ,  ,
,  add
Show full author list
add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection, Library Construction, and Sequencing Methods
2.2. Viral Genome Assembly: Quality Check and Mapping of the Reads
2.3. Sequence Selection from GISAID
2.4. Minimum Spanning Network (MSN)
2.5. Phylogenetic Analysis
2.6. Metadata Collection
3. Results
3.1. Viral Haplotypes Circulating in Vo’
3.2. Vo’ Haplotypes in Europe
3.3. Intra-Host Viral Evolution
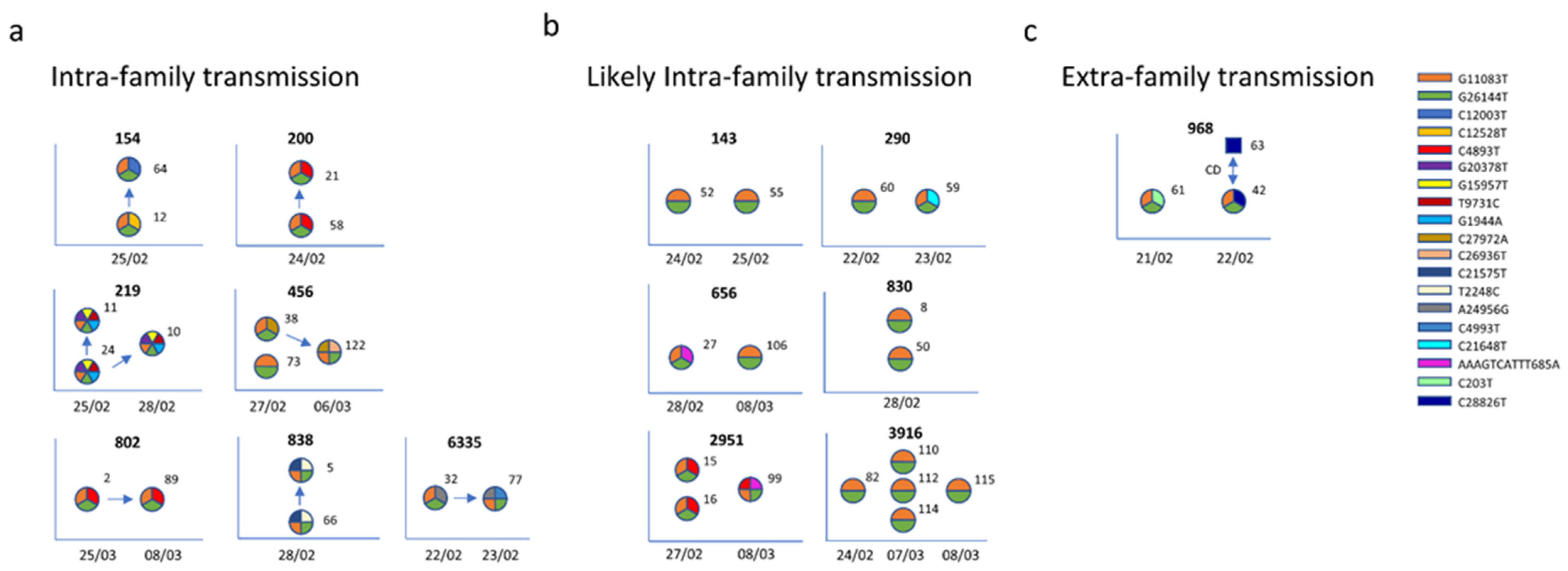
3.4. Within-Household Variability
3.5. Transmission Chain Reconstruction
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Intra-Family Transmission, a Detailed Description
References
- WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int (accessed on 10 February 2022).
- Islam, M.R.; Hoque, M.N.; Rahman, M.S.; Alam, A.S.M.R.U.; Akther, M.; Puspo, J.A.; Akter, S.; Sultana, M.; Crandall, K.A.; Hossain, M.A. Genome-Wide Analysis of SARS-CoV-2 Virus Strains Circulating Worldwide Implicates Heterogeneity. Sci. Rep. 2020, 10, 14004. [Google Scholar] [CrossRef]
- Laamarti, M.; Alouane, T.; Kartti, S.; Chemao-Elfihri, M.W.; Hakmi, M.; Essabbar, A.; Laamarti, M.; Hlali, H.; Bendani, H.; Boumajdi, N.; et al. Large Scale Genomic Analysis of 3067 SARS-CoV-2 Genomes Reveals a Clonal Geo-Distribution and a Rich Genetic Variations of Hotspots Mutations. PLoS ONE 2020, 15, e0240345. [Google Scholar] [CrossRef] [PubMed]
- Roy, C.; Mandal, S.M.; Mondal, S.K.; Mukherjee, S.; Mapder, T.; Ghosh, W.; Chakraborty, R. Trends of Mutation Accumulation across Global SARS-CoV-2 Genomes: Implications for the Evolution of the Novel Coronavirus. Genomics 2020, 112, 5331–5342. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence That D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.; Plante, K.S.; Plante, J.A.; Xie, X.; Zhang, X.; Ku, Z.; An, Z.; Scharton, D.; Schindewolf, C.; et al. The N501Y Spike Substitution Enhances SARS-CoV-2 Infection and Transmission. Nature 2022, 602, 294–299. [Google Scholar] [CrossRef]
- Wang, P.; Nair, M.S.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; et al. Antibody Resistance of SARS-CoV-2 Variants B.1.351 and B.1.1.7. Nature 2021, 593, 130–135. [Google Scholar] [CrossRef]
- Tao, K.; Tzou, P.L.; Nouhin, J.; Gupta, R.K.; de Oliveira, T.; Kosakovsky Pond, S.L.; Fera, D.; Shafer, R.W. The Biological and Clinical Significance of Emerging SARS-CoV-2 Variants. Nat. Rev. Genet. 2021, 22, 757–773. [Google Scholar] [CrossRef]
- Karim, S.S.A.; Karim, Q.A. Omicron SARS-CoV-2 Variant: A New Chapter in the COVID-19 Pandemic. Lancet 2021, 398, 2126–2128. [Google Scholar] [CrossRef]
- Lavezzo, E.; Franchin, E.; Ciavarella, C.; Cuomo-Dannenburg, G.; Barzon, L.; Del Vecchio, C.; Rossi, L.; Manganelli, R.; Loregian, A.; Navarin, N. Suppression of a SARS-CoV-2 Outbreak in the Italian Municipality of Vo’. Nature 2020, 584, 425–429. [Google Scholar] [CrossRef]
- Gittelman, R.M.; Lavezzo, E.; Snyder, T.M.; Zahid, H.J.; Elyanow, R.; Dalai, S.; Kirsch, I.; Baldo, L.; Manuto, L.; Franchin, E.; et al. Diagnosis and Tracking of SARS-CoV-2 Infection By T-Cell Receptor Sequencing. 2021. Available online: https://www.medrxiv.org/content/10.1101/2020.11.09.20228023v2 (accessed on 10 February 2022).
- Dorigatti, I.; Lavezzo, E.; Manuto, L.; Ciavarella, C.; Pacenti, M.; Boldrin, C.; Cattai, M.; Saluzzo, F.; Franchin, E.; Del Vecchio, C.; et al. SARS-CoV-2 Antibody Dynamics and Transmission from Community-Wide Serological Testing in the Italian Municipality of Vo’. Nat. Commun. 2021, 12, 4383. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for Clustering the next-Generation Sequencing Data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-Joining Networks for Inferring Intraspecific Phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- CDC Health Departments. Available online: https://www.cdc.gov/coronavirus/2019-ncov/php/contact-tracing/keyinfo.html (accessed on 10 February 2022).
- Elbe, S.; Buckland-Merrett, G. Data, Disease and Diplomacy: GISAID’s Innovative Contribution to Global Health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Capobianchi, M.R.; Rueca, M.; Messina, F.; Giombini, E.; Carletti, F.; Colavita, F.; Castilletti, C.; Lalle, E.; Bordi, L.; Vairo, F.; et al. Molecular Characterization of SARS-CoV-2 from the First Case of COVID-19 in Italy. Clin. Microbiol. Infect. 2020, 26, 954–956. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Bianco, L.; Moser, M.; Silverj, A.; Micheletti, D.; Lorenzin, G.; Collini, L.; Barbareschi, M.; Lanzafame, P.; Segata, N.; Pindo, M.; et al. On the Origin and Propagation of the Covid-19 Outbreak in the Italian Province of Trento, a Tourist Region of Northern Italy. Viruses.
- Lai, A.; Bergna, A.; Caucci, S.; Clementi, N.; Vicenti, I.; Dragoni, F.; Cattelan, A.M.; Menzo, S.; Pan, A.; Callegaro, A.; et al. Molecular Tracing of SARS-CoV-2 in Italy in the First Three Months of the Epidemic. Viruses 2020, 12, 798. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, F.; Snyder, G.A.; Giovanetti, M.; Angeletti, S.; Gallo, R.C.; Ciccozzi, M.; Zella, D. Emerging of a SARS-CoV-2 Viral Strain with a Deletion in Nsp1. J. Transl. Med. 2020, 18, 329. [Google Scholar] [CrossRef]
- van Dorp, L.; Acman, M.; Richard, D.; Shaw, L.P.; Ford, C.E.; Ormond, L.; Owen, C.J.; Pang, J.; Tan, C.C.S.; Boshier, F.A.T.; et al. Emergence of Genomic Diversity and Recurrent Mutations in SARS-CoV-2. Infect. Genet. Evol. 2020, 83, 104351. [Google Scholar] [CrossRef] [PubMed]
- Annavajhala, M.K.; Mohri, H.; Wang, P.; Nair, M.; Zucker, J.E.; Sheng, Z.; Gomez-Simmonds, A.; Kelley, A.L.; Tagliavia, M.; Huang, Y.; et al. Emergence and Expansion of SARS-CoV-2 B.1.526 after Identification in New York. Nature 2021, 597, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Voss, J.D.; Skarzynski, M.; McAuley, E.M.; Maier, E.J.; Gibbons, T.; Fries, A.C.; Chapleau, R.R. Variants in SARS-CoV-2 Associated with Mild or Severe Outcome. Evol. Med. Public Health 2021, 9, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Benvenuto, D.; Angeletti, S.; Giovanetti, M.; Bianchi, M.; Pascarella, S.; Cauda, R.; Ciccozzi, M.; Cassone, A. Evolutionary Analysis of SARS-CoV-2: How Mutation of Non-Structural Protein 6 (NSP6) Could Affect Viral Autophagy. J. Infect. 2020, 81, e24–e27. [Google Scholar] [CrossRef] [PubMed]
- Zepeda-Cervantes, J.; Martínez-Flores, D.; Ramírez-Jarquín, J.O.; Tecalco-Cruz, Á.C.; Alavez-Pérez, N.S.; Vaca, L.; Sarmiento-Silva, R.E. Implications of the Immune Polymorphisms of the Host and the Genetic Variability of SARS-CoV-2 in the Development of COVID-19. Viruses 2022, 14, 94. [Google Scholar] [CrossRef] [PubMed]
- Zandi, M.; Soltani, S.; Sanami, S.; Rasooli, A. Spike Protein Mutations and the Effects on SARS-CoV-2 Pathogenesis. J. Cell. Mol. Anesth. 2021, 6, 148–153. [Google Scholar] [CrossRef]
- Guo, E.; Guo, H. CD8 T Cell Epitope Generation toward the Continually Mutating SARS-CoV-2 Spike Protein in Genetically Diverse Human Population: Implications for Disease Control and Prevention. PLoS ONE 2020, 15, e0239566. [Google Scholar] [CrossRef]
- Pachetti, M.; Marini, B.; Giudici, F.; Benedetti, F.; Angeletti, S.; Ciccozzi, M.; Masciovecchio, C.; Ippodrino, R.; Zella, D. Impact of Lockdown on Covid-19 Case Fatality Rate and Viral Mutations Spread in 7 Countries in Europe and North America. J. Transl. Med. 2020, 18, 338. [Google Scholar] [CrossRef]
- Gill, M.; Gray, M. Mass Testing for Covid-19 in the UK. BMJ 2020, 371, m4436. [Google Scholar] [CrossRef]
- Gudbjartsson, D.F.; Helgason, A.; Jonsson, H.; Magnusson, O.T.; Melsted, P.; Norddahl, G.L.; Saemundsdottir, J.; Sigurdsson, A.; Sulem, P.; Agustsdottir, A.B.; et al. Spread of SARS-CoV-2 in the Icelandic Population. N. Engl. J. Med. 2020, 382, 2302–2315. [Google Scholar] [CrossRef]
- Lai, A.; Bergna, A.; Toppo, S.; Morganti, M.; Menzo, S.; Ghisetti, V.; Bruzzone, B.; Codeluppi, M.; Fiore, V.; Rullo, E.V.; et al. Evolutionary Dynamics of SARS-CoV-2 in Space and Time During the First Phase of the Epidemic in Italy. Preprints 2021. [Google Scholar] [CrossRef]
- Hensley, M.K.; Bain, W.G.; Jacobs, J.; Nambulli, S.; Parikh, U.; Cillo, A.; Staines, B.; Heaps, A.; Sobolewski, M.D.; Rennick, L.J.; et al. Intractable Coronavirus Disease 2019 (COVID-19) and Prolonged Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Replication in a Chimeric Antigen Receptor-Modified T-Cell Therapy Recipient: A Case Study. Clin. Infect. Dis. 2021, 73, e815–e821. [Google Scholar] [CrossRef] [PubMed]
- Kemp, S.A.; Collier, D.A.; Datir, R.P.; Ferreira, I.A.T.M.; Gayed, S.; Jahun, A.; Hosmillo, M.; Rees-Spear, C.; Mlcochova, P.; Lumb, I.U.; et al. SARS-CoV-2 Evolution during Treatment of Chronic Infection. Nature 2021, 592, 277–282. [Google Scholar] [CrossRef]
- Choi, B.; Choudhary, M.C.; Regan, J.; Sparks, J.A.; Padera, R.F.; Qiu, X.; Solomon, I.H.; Kuo, H.-H.; Boucau, J.; Bowman, K.; et al. Persistence and Evolution of SARS-CoV-2 in an Immunocompromised Host. N. Engl. J. Med. 2020, 383, 2291–2293. [Google Scholar] [CrossRef] [PubMed]
- Khatamzas, E.; Rehn, A.; Muenchhoff, M.; Hellmuth, J.; Gaitzsch, E.; Weiglein, T.; Georgi, E.; Scherer, C.; Stecher, S.; Weigert, O.; et al. Emergence of Multiple SARS-CoV-2 Mutations in an Immunocompromised Host. 2021; Available online: https://www.medrxiv.org/content/10.1101/2021.01.10.20248871v1 (accessed on 10 February 2022).
- Lythgoe, K.A.; Hall, M.; Ferretti, L.; de Cesare, M.; MacIntyre-Cockett, G.; Trebes, A.; Andersson, M.; Otecko, N.; Wise, E.L.; Moore, N.; et al. SARS-CoV-2 within-Host Diversity and Transmission. Science 2021, 372, eabg0821. [Google Scholar] [CrossRef]
- Voloch, C.M.; da Silva Francisco Jr, R.; de Almeida, L.G.P.; Brustolini, O.J.; Cardoso, C.C.; Gerber, A.L.; Guimarães, A.P. de C.; Leitão, I. de C.; Mariani, D.; Ota, V.A.; et al. Intra-Host Evolution during SARS-CoV-2 Prolonged Infection. Virus Evol. 2021, 7. [Google Scholar] [CrossRef]
- Wang, R.; Hozumi, Y.; Zheng, Y.-H.; Yin, C.; Wei, G.-W. Host Immune Response Driving SARS-CoV-2 Evolution. Viruses 2020, 12, 1095. [Google Scholar] [CrossRef]
- Azgari, C.; Kilinc, Z.; Turhan, B.; Circi, D.; Adebali, O. The Mutation Profile of SARS-CoV-2 Is Primarily Shaped by the Host Antiviral Defense. Viruses 2021, 13, 394. [Google Scholar] [CrossRef]
- Garry, M.; Hope, L.; Zajac, R.; Verrall, A.J.; Robertson, J.M. Contact Tracing: A Memory Task With Consequences for Public Health. Perspect. Psychol. Sci. 2021, 16, 175–187. [Google Scholar] [CrossRef]
- Pearce, N.; Lawlor, D.A.; Brickley, E.B. Comparisons between Countries Are Essential for the Control of COVID-19. Int. J. Epidemiol. 2020, 49, 1059–1062. [Google Scholar] [CrossRef]
- Peto, J. Covid-19 Mass Testing Facilities Could End the Epidemic Rapidly. BMJ 2020, 368, m1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosetti, P.; Kiem, C.T.; Yazdanpanah, Y.; Fontanet, A.; Lina, B.; Colizza, V.; Cauchemez, S. Impact of Mass Testing during an Epidemic Rebound of SARS-CoV-2: A Modelling Study Using the Example of France. Eurosurveillance 2021, 26, 2001978. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SUBJECTS | NOTES | |
|---|---|---|
| HOUSEHOLDS | SR_38, SR_122, SR_64, SR_12, and SR_77 | Subjects infected by family members carrying a different haplotype |
| HAPLOTYPES | SR_56 and SR_99 | Subjects carrying a haplotype that evolved from a subtype of the AH |
| SYMPTOM ONSET DATES | SR_65, SR_61, and SR_30 | First subjects contracting the infection, likely infected with the AH |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manuto, L.; Grazioli, M.; Spitaleri, A.; Fontana, P.; Bianco, L.; Bertolotti, L.; Bado, M.; Mazzotti, G.; Bianca, F.; Onelia, F.; et al. Rapid SARS-CoV-2 Intra-Host and Within-Household Emergence of Novel Haplotypes. Viruses 2022, 14, 399. https://doi.org/10.3390/v14020399
Manuto L, Grazioli M, Spitaleri A, Fontana P, Bianco L, Bertolotti L, Bado M, Mazzotti G, Bianca F, Onelia F, et al. Rapid SARS-CoV-2 Intra-Host and Within-Household Emergence of Novel Haplotypes. Viruses. 2022; 14(2):399. https://doi.org/10.3390/v14020399
Chicago/Turabian StyleManuto, Laura, Marco Grazioli, Andrea Spitaleri, Paolo Fontana, Luca Bianco, Luigi Bertolotti, Martina Bado, Giorgia Mazzotti, Federico Bianca, Francesco Onelia, and et al. 2022. "Rapid SARS-CoV-2 Intra-Host and Within-Household Emergence of Novel Haplotypes" Viruses 14, no. 2: 399. https://doi.org/10.3390/v14020399
APA StyleManuto, L., Grazioli, M., Spitaleri, A., Fontana, P., Bianco, L., Bertolotti, L., Bado, M., Mazzotti, G., Bianca, F., Onelia, F., Lorenzin, G., Simeoni, F., Lazarevic, D., Franchin, E., Vecchio, C. D., Dorigatti, I., Tonon, G., Cirillo, D. M., Lavezzo, E., ... Toppo, S. (2022). Rapid SARS-CoV-2 Intra-Host and Within-Household Emergence of Novel Haplotypes. Viruses, 14(2), 399. https://doi.org/10.3390/v14020399