
Epigenomic and Proteomic Changes in Fetal Spleens Persistently Infected with Bovine Viral Diarrhea Virus: Repercussions for the Developing Immune System, Bone, Brain, and Heart


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals, Viral Infections, and Fetal Collections
2.2. Reduced Representation Bisulfite Sequencing
2.3. Methylation Bioinformatics and Pathway Analysis
2.4. Protein Sample Preparation
2.5. Liquid Chromatography and Mass Spectrometry
2.6. Proteomics Data Analysis and Instrument Quality Control
3. Results
3.1. Reduced Representation Bisulfite Sequencing
3.2. Proteomics
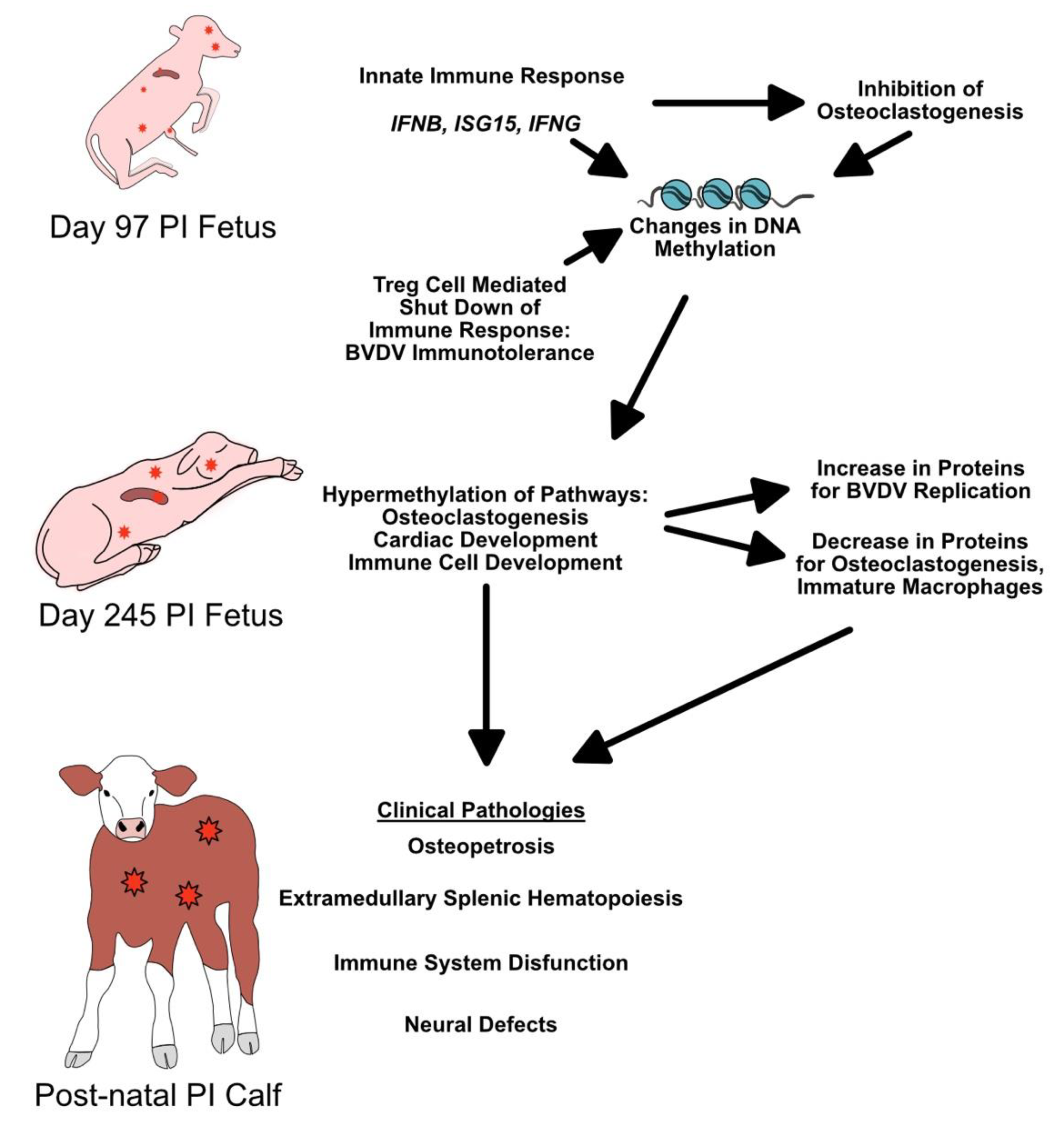
4. Discussion
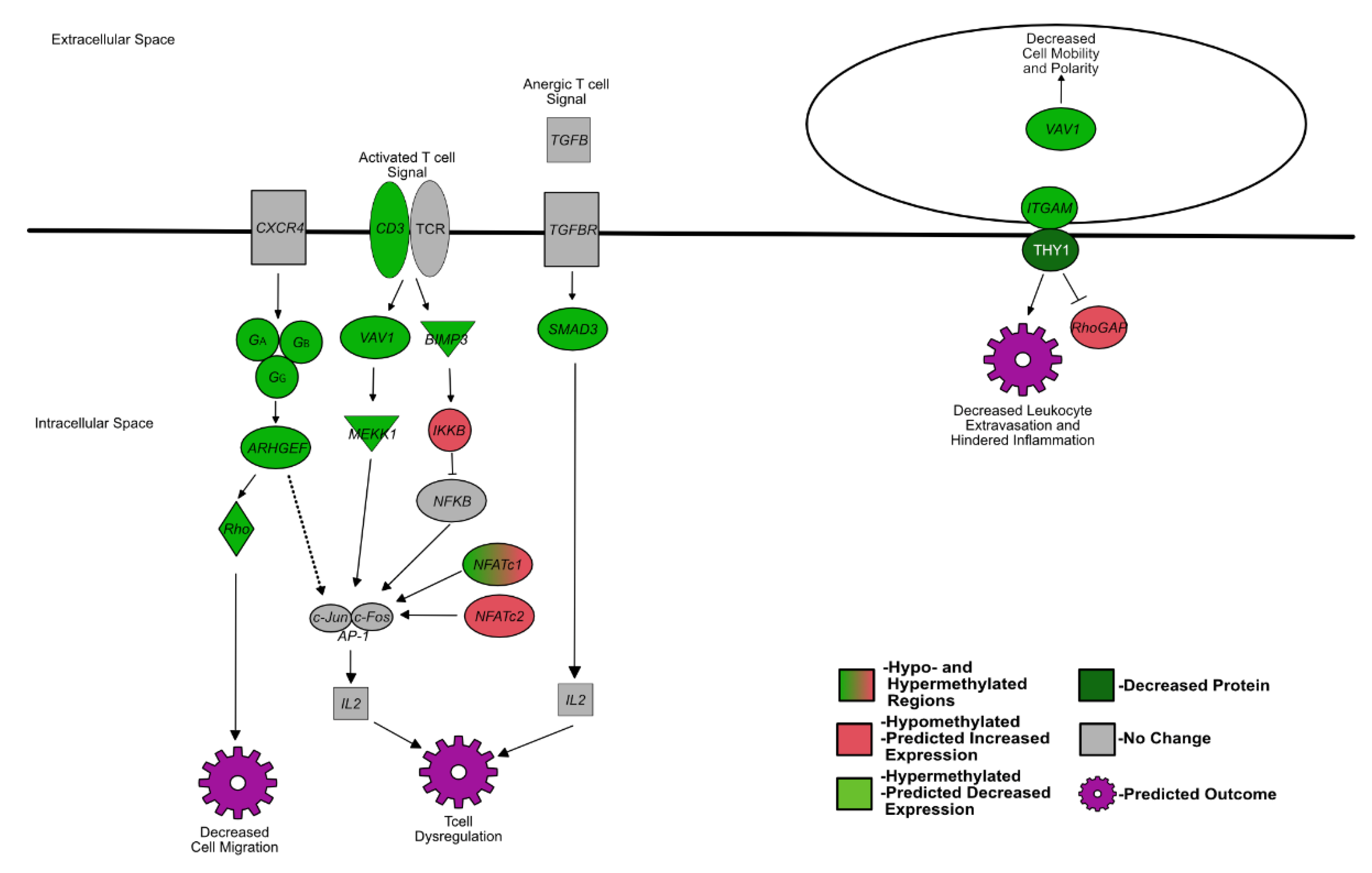
4.1. Fetal BVDV PI Alters the Expression of Genes That Influence Lymphocyte Development
4.2. BVDV Fetal Infection Alters Osteoclastogenesis Resulting in Extramedullary Splenic Hematopoiesis
4.3. Methylation Patterns and Protein Expression in the PI Fetal Spleen Are Related to Neural Defects in PI Fetuses
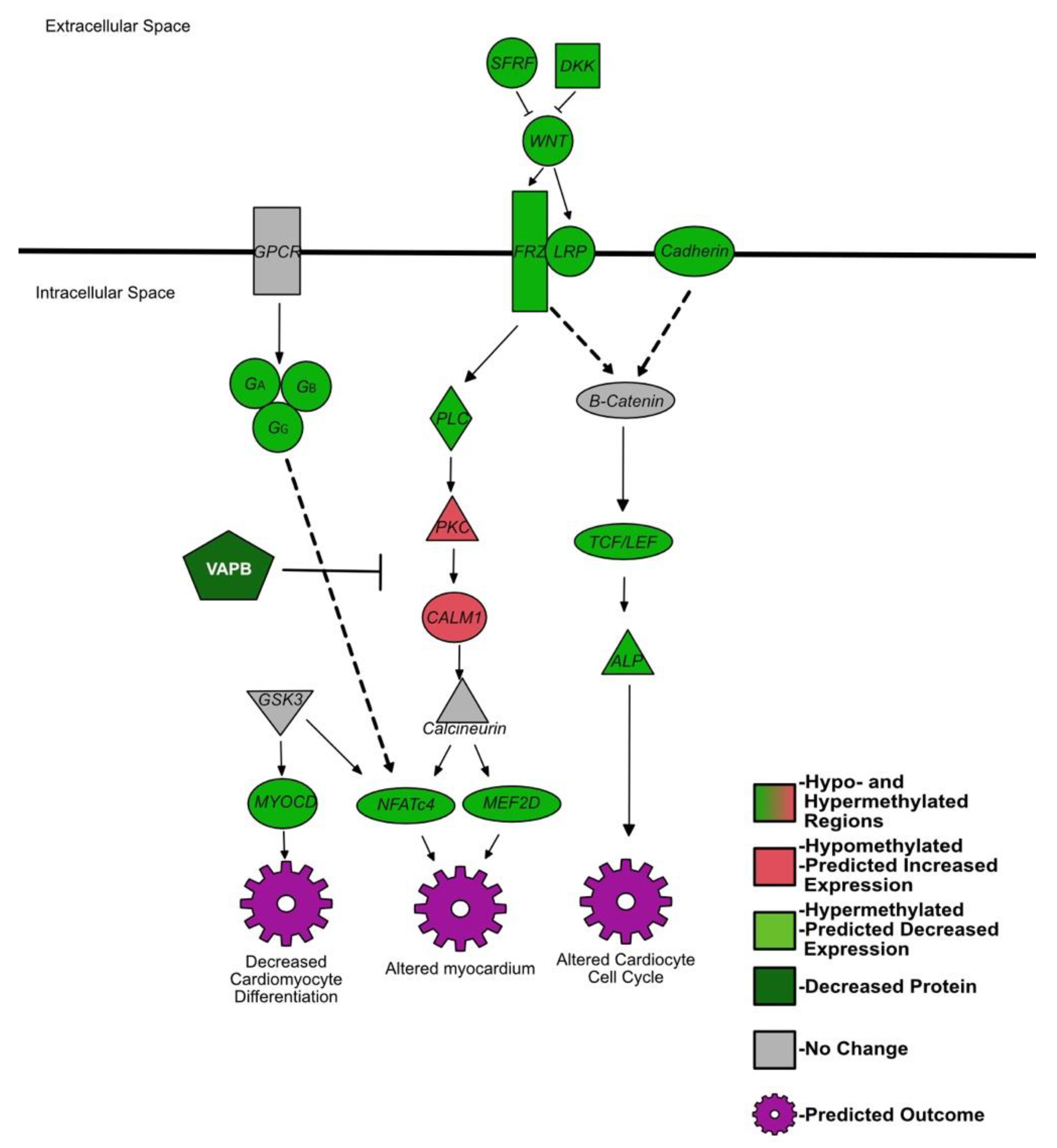
4.4. Genes Affecting Cardiac Development Are Hypermethylated in the Spleen of BVDV PI Fetuses
4.5. BVDV Replication and Immune Evasion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Olafson, P.; Maccallum, A.D.; Fox, F.H. An apparently new transmissible disease of cattle. Cornell Veter. 1946, 36, 205–213. [Google Scholar]
- Ridpath, J.F. Bovine Viral Diarrhea Virus: Global Status. Vet. Clin. N. Am. Food Anim. Pract. 2010, 26, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Kalaycioglu, A. Bovine viral diarrhoea virus (BVDV) diversity and vaccination. A review. Veter. Q. 2007, 29, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Gillespie, J.H. Propagation of virus diarrhea virus of cattle in tissue culture. Am. J. Veter. Res. 1957, 18, 952–953. [Google Scholar]
- Collett, M.S.; Moennig, V.; Horzinek, M.C. Recent Advances in Pestivirus Research. J. Gen. Virol. 1989, 70, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Underdahl, N.R.; Grace, O.D.; Hoerlein, A.B. Cultivation in Tissue-Culture of Cytopathogenic Agent from Bovine Mucosal Disease. Exp. Biol. Med. 1957, 94, 795–797. [Google Scholar] [CrossRef]
- Ridpath, J.; Bolin, S.; Dubovi, E.J. Segregation of Bovine Viral Diarrhea Virus into Genotypes. Virology 1994, 205, 66–74. [Google Scholar] [CrossRef]
- Donis, R.O. Molecular Biology of Bovine Viral Diarrhea Virus and its Interactions with the Host. Veter. Clin. N. Am. Food Anim. Pract. 1995, 11, 393–423. [Google Scholar] [CrossRef]
- Kupfermann, H.; Thiel, H.J.; Dubovi, E.J.; Meyers, G. Bovine viral diarrhea virus: Characterization of a cytopathogenic defective interfering particle with two internal deletions. J. Virol. 1996, 70, 8175. [Google Scholar] [CrossRef] [Green Version]
- Fritzemeier, J.; Haas, L.; Liebler, E.; Moennig, V.; Greiser-Wilke, I. The development of early vs. late onset mucosal disease is a consequence of two different pathogenic mechanisms. Arch. Virol. 1997, 142, 1335–1350. [Google Scholar] [CrossRef]
- Tautz, N.; Thiel, H.-J. Cytopathogenicity of pestiviruses: Cleavage of bovine viral diarrhea virus NS2-3 has to occur at a defined position to allow viral replication. Arch. Virol. 2003, 148, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.C. The Clinical Manifestations of Bovine Viral Diarrhea Infection. Veter. Clin. N. Am. Food Anim. Pract. 1995, 11, 425–445. [Google Scholar] [CrossRef]
- Bielefeldt-Ohmann, H. The Pathologies of Bovine Viral Diarrhea Virus Infection: A Window on the Pathogenesis. Vet. Clin. N. Am. Food Anim. Pract. 1995, 11, 447–476. [Google Scholar] [CrossRef]
- Moennig, V.; Liess, B. Pathogenesis of Intrauterine Infections with Bovine Viral Diarrhea Virus. Veter. Clin. N. Am. Food Anim. Pract. 1995, 11, 477–487. [Google Scholar] [CrossRef]
- McClurkin, A.W.; Littledike, E.T.; Cutlip, R.C.; Frank, G.H.; Coria, M.F.; Bolin, S.R. Production of cattle immunotolerant to bovine viral diarrhea virus. Can. J. Comp. Med. Rev. Can. Med. Comp. 1984, 48, 156–161. [Google Scholar]
- Brownlie, J.; Clarke, M.C.; Howard, C.J. Experimental production of fatal mucosal disease in cattle. Veter. Rec. 1984, 114, 535–536. [Google Scholar] [CrossRef] [PubMed]
- Scruggs, D.W.; Fleming, S.A.; Maslin, W.R.; Wayne, G.A. Osteopetrosis, Anemia, Thrombocytopenia, and Marrow Necrosis in Beef Calves Naturally Infected with Bovine Virus Diarrhea Virus. J. Veter. Diagn. Investig. 1995, 7, 555–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiess, A.; Hilbe, M.; Sterr, K.; Reiser, M.; Matis, U.; Nuss, K. Transient benign osteopetrosis in a calf persistently infected with bovine virus diarrhoea virus. Veter. Comp. Orthop. Traumatol. 2005, 18, 100–104. [Google Scholar] [CrossRef]
- Done, J.T.; Terlecki, S.; Richardson, C.; Harkness, J.W.; Sands, J.J.; Patterson, D.S.; Sweasey, D.; Shaw, I.G.; Winkler, C.E.; Duffell, S.J. Bovine virus diarrhoea-mucosal disease virus: Pathogenicity for the fetal calf following maternal infection. Veter. Rec. 1980, 106, 473–479. [Google Scholar] [CrossRef]
- Webb, B.T.; Norrdin, R.W.; Smirnova, N.P.; Van Campen, H.; Weiner, C.M.; Antoniazzi, A.Q.; Bielefeldt-Ohmann, H.; Hansen, T.R. Bovine Viral Diarrhea Virus Cyclically Impairs Long Bone Trabecular Modeling in Experimental Persistently Infected Fetuses. Veter. Pathol. 2012, 49, 930–940. [Google Scholar] [CrossRef]
- Shoemaker, M.L.; Smirnova, N.P.; Bielefeldt-Ohmann, H.; Austin, K.J.; van Olphen, A.; Clapper, J.A.; Hansen, T.R. Differential Expression of the Type I Interferon Pathway during Persistent and Transient Bovine Viral Diarrhea Virus Infection. J. Interf. Cytokine Res. 2009, 29, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.R.; Smirnova, N.P.; Van Campen, H.; Shoemaker, M.L.; Ptitsyn, A.; Bielefeldt-Ohmann, H. Maternal and Fetal Response to Fetal Persistent Infection with Bovine Viral Diarrhea Virus. Am. J. Reprod. Immunol. 2010, 64, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, N.P.; Webb, B.T.; Bielefeldt-Ohmann, H.; Van Campen, H.; Antoniazzi, A.Q.; Morarie, S.E.; Hansen, T.R. Development of fetal and placental innate immune responses during establishment of persistent infection with bovine viral diarrhea virus. Virus Res. 2012, 167, 329–336. [Google Scholar] [CrossRef]
- Georges, H.M.; Knapek, K.J.; Bielefeldt-Ohmann, H.; Van Campen, H.; Hansen, T.R. Attenuated lymphocyte activation leads to the development of immunotolerance in bovine fetuses persistently infected with BVDV. Biol. Reprod. 2020, 103, 560–571. [Google Scholar] [CrossRef] [PubMed]
- Knapek, K.J.; Georges, H.; Van Campen, H.; Bishop, J.V.; Bielefeldt-Ohmann, H.; Smirnova, N.P.; Hansen, T.R. Fetal Lymphoid Organ Immune Responses to Transient and Persistent Infection with Bovine Viral Diarrhea Virus. Viruses 2020, 12, 816. [Google Scholar] [CrossRef]
- Webb, B.T.; McGilvray, K.C.; Smirnova, N.P.; Hansen, T.R.; Norrdin, R.W. Effects of in utero pestivirus infection on bovine fetal bone geometry, biomechanical properties and composition. Veter. J. 2013, 198, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Bielefeldt-Ohmann, H.; Smirnova, N.P.; Tolnay, A.-E.; Webb, B.T.; Antoniazzi, A.Q.; van Campen, H.; Hansen, T.R. Neuro-invasion by a ‘Trojan Horse’ strategy and vasculopathy during intrauterine flavivirus infection. Int. J. Exp. Pathol. 2012, 93, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, N.P.; Bielefeldt-Ohmann, H.; Van Campen, H.; Austin, K.J.; Han, H.; Montgomery, D.L.; Shoemaker, M.L.; van Olphen, A.L.; Hansen, T.R. Acute non-cytopathic bovine viral diarrhea virus infection induces pronounced type I interferon response in pregnant cows and fetuses. Virus Res. 2008, 132, 49–58. [Google Scholar] [CrossRef]
- Smirnova, N.P.; Webb, B.T.; McGill, J.L.; Schaut, R.G.; Bielefeldt-Ohmann, H.; Van Campen, H.; Sacco, R.E.; Hansen, T.R. Induction of interferon-gamma and downstream pathways during establishment of fetal persistent infection with bovine viral diarrhea virus. Virus Res. 2014, 183, 95–106. [Google Scholar] [CrossRef]
- Peterhans, E.; Jungi, T.W.; Schweizer, M. BVDV and innate immunity. Biologicals 2003, 31, 107–112. [Google Scholar] [CrossRef]
- Schweizer, M.; Peterhans, E. Noncytopathic Bovine Viral Diarrhea Virus Inhibits Double-Stranded RNA-Induced Apoptosis and Interferon Synthesis. J. Virol. 2001, 75, 4692–4698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akalin, A.; Franke, V.; Vlahoviček, K.; Mason, C.E.; Schübeler, D. Genomation: A toolkit to summarize, annotate and visualize genomic intervals. Bioinformatics 2015, 31, 1127–1129. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Friedman, M.S.; Shedden, K.; Hankenson, K.D.; Woolf, P.J. GAGE: Generally applicable gene set enrichment for pathway analysis. BMC Bioinform. 2009, 10, 161. [Google Scholar] [CrossRef] [Green Version]
- Georges, H.M. Hanahm1/methyl_seq, 1; Github. 2022. Available online: http://github.com/hanahm1/methyl_seq (accessed on 19 February 2022).
- Schauer, K.; Freund, D.; Prenni, J.; Curthoys, N.P. Proteomic profiling and pathway analysis of the response of rat renal proximal convoluted tubules to metabolic acidosis. Am. J. Physiol. Physiol. 2013, 305, F628–F640. [Google Scholar] [CrossRef] [Green Version]
- Scopes, R. Measurement of protein by spectrophotometry at 205 nm. Anal. Biochem. 1974, 59, 277–282. [Google Scholar] [CrossRef]
- Keller, A.; Nesvizhskii, A.I.; Kolker, E.; Aebersold, R. Empirical Statistical Model to Estimate the Accuracy of Peptide Identifications Made by MS/MS and Database Search. Anal. Chem. 2002, 74, 5383–5392. [Google Scholar] [CrossRef]
- Searle, B.C.; Turner, M.; Nesvizhskii, A. Improving Sensitivity by Probabilistically Combining Results from Multiple MS/MS Search Methodologies. J. Proteome Res. 2008, 7, 245–253. [Google Scholar] [CrossRef]
- Käll, L.; Storey, J.; MacCoss, M.J.; Noble, W.S. Assigning Significance to Peptides Identified by Tandem Mass Spectrometry Using Decoy Databases. J. Proteome Res. 2007, 7, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Nesvizhskii, A.I.; Keller, A.; Kolker, E.; Aebersold, R. A Statistical Model for Identifying Proteins by Tandem Mass Spectrometry. Anal. Chem. 2003, 75, 4646–4658. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Sadygov, R.G.; Yates, J.R. A Model for Random Sampling and Estimation of Relative Protein Abundance in Shotgun Proteomics. Anal. Chem. 2004, 76, 4193–4201. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Lin, J.-X.; Leonard, W.J. Interleukin-2 at the Crossroads of Effector Responses, Tolerance, and Immunotherapy. Immunity 2013, 38, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Cheng, G.; Yu, A.; Malek, T.R. T-cell tolerance and the multi-functional role of IL-2R signaling in T-regulatory cells. Immunol. Rev. 2011, 241, 63–76. [Google Scholar] [CrossRef] [Green Version]
- Busse, D.; de la Rosa, M.; Hobiger, K.; Thurley, K.; Flossdorf, M.; Scheffold, A.; Hofer, T. Competing feedback loops shape IL-2 signaling between helper and regulatory T lymphocytes in cellular microenvironments. Proc. Natl. Acad. Sci. USA 2010, 107, 3058–3063. [Google Scholar] [CrossRef] [Green Version]
- Sadlack, B.; Löhler, J.; Schorle, H.; Klebb, G.; Haber, H.; Sickel, E.; Noelle, R.J.; Horak, I. Generalized autoimmune disease in interleukin-2-deficient mice is triggered by an uncontrolled activation and proliferation of CD4+ T cells. Eur. J. Immunol. 1995, 25, 3053–3059. [Google Scholar] [CrossRef]
- Malek, T.R.; Yu, A.; Vincek, V.; Scibelli, P.; Kong, L. CD4 Regulatory T Cells Prevent Lethal Autoimmunity in IL-2Rβ-Deficient Mice: Implications for the Nonredundant Function of IL-2. Immunity 2002, 17, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Schubert, K.; Polte, T.; Bönisch, U.; Schader, S.; Holtappels, R.; Hildebrandt, G.; Lehmann, J.; Simon, J.C.; Anderegg, U.; Saalbach, A. Thy-1 (CD90) regulates the extravasation of leukocytes during inflammation. Eur. J. Immunol. 2011, 41, 645–656. [Google Scholar] [CrossRef]
- Fischer, R.S.; Fritz-Six, K.L.; Fowler, V.M.; Arakawa, Y.; Bito, H.; Furuyashiki, T.; Tsuji, T.; Takemoto-Kimura, S.; Kimura, K.; Nozaki, K.; et al. Pointed-end capping by tropomodulin3 negatively regulates endothelial cell motility. J. Cell Biol. 2003, 161, 371–380. [Google Scholar] [CrossRef] [Green Version]
- Helal, M.; Hoshino, Y.; Takagi, S.; Tajima, M. C-X-C chemokine receptor type 4 and cytokine expressions in cows of a dairy herd with high prevalence of calves persistently infected with bovine viral diarrhea virus. Jpn. J. Veter. Res. 2013, 61. [Google Scholar]
- Pozzobon, T.; Goldoni, G.; Viola, A.; Molon, B. CXCR4 signaling in health and disease. Immunol. Lett. 2016, 177, 6–15. [Google Scholar] [CrossRef]
- Huang, L.J.-S.; Durick, K.; Weiner, J.; Chun, J.; Taylor, S.S. D-AKAP2, a novel protein kinase A anchoring protein with a putative RGS domain. Proc. Natl. Acad. Sci. USA 1997, 94, 11184–11189. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Dong, Y.; Tu, K.; Wang, W. Proteomics analysis reveals the interleukin-35-dependent regulatory mechanisms affecting CD8+ T-cell functions. Cell. Immunol. 2019, 348, 104022. [Google Scholar] [CrossRef] [PubMed]
- Olferiev, M.; Jacek, E.; Kirou, K.A.; Crow, M.K. Novel molecular signatures in mononuclear cell populations from patients with systemic lupus erythematosus. Clin. Immunol. 2016, 172, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Tsukasaki, M.; Takayanagi, H. Osteoimmunology: Evolving concepts in bone–immune interactions in health and disease. Nat. Rev. Immunol. 2019, 19, 626–642. [Google Scholar] [CrossRef] [PubMed]
- You, X.; Bian, C.; Zan, Q.; Xu, X.; Liu, X.; Chen, J.; Wang, J.; Qiu, Y.; Li, W.; Zhang, X.; et al. Mudskipper genomes provide insights into the terrestrial adaptation of amphibious fishes. Nat. Commun. 2014, 5, 5594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arron, J.R.; Choi, Y. Bone versus immune system. Nature 2000, 408, 535–536. [Google Scholar] [CrossRef]
- Takayanagi, H.; Ogasawara, K.; Hida, S.; Chiba, T.; Murata, S.; Sato, K.; Takaoka, A.; Yokochi, T.; Oda, H.; Tanaka, K.; et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-γ. Nature 2000, 408, 600–605. [Google Scholar] [CrossRef]
- Okamoto, K.; Nakashima, T.; Shinohara, M.; Negishi-Koga, T.; Komatsu, N.; Terashima, A.; Sawa, S.; Nitta, T.; Takayanagi, H. Osteoimmunology: The Conceptual Framework Unifying the Immune and Skeletal Systems. Physiol. Rev. 2017, 97, 1295–1349. [Google Scholar] [CrossRef]
- Asagiri, M.; Sato, K.; Usami, T.; Ochi, S.; Nishina, H.; Yoshida, H.; Morita, I.; Wagner, E.F.; Mak, T.W.; Serfling, E.; et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J. Exp. Med. 2005, 202, 1261–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takayanagi, H. Osteoimmunology and the effects of the immune system on bone. Nat. Rev. Rheumatol. 2009, 5, 667–676. [Google Scholar] [CrossRef]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.-I.; et al. Induction and Activation of the Transcription Factor NFATc1 (NFAT2) Integrate RANKL Signaling in Terminal Differentiation of Osteoclasts. Dev. Cell 2002, 3, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Gerritsen, E.J.; Vossen, J.M.; van Loo, I.H.; Hermans, J.; Helfrich, M.H.; Griscelli, C.; Fischer, A. Autosomal recessive osteopetrosis: Variability of findings at diagnosis and during the natural course. Pediatrics 1994, 93, 247–253. [Google Scholar] [CrossRef]
- Tolar, J.; Teitelbaum, S.L.; Orchard, P.J. Osteopetrosis. N. Engl. J. Med. 2004, 351, 2839–2849. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.J.; Carey, L.S. Rubella embryopathy: Roentgenologic features. Am. J. Roentgenol. 1966, 97, 92–99. [Google Scholar] [CrossRef] [Green Version]
- Graham, C.B.; Thal, A.; Wassum, C.S. Rubella-Like Bone Changes in Congenital Cytomegalic Inclusion Disease. Radiology 1970, 94, 39–43. [Google Scholar] [CrossRef]
- Kopelman, A.E.; Halsted, C.C.; Minnefor, A.B. Osteomalacia and spontaneous fractures in twins with congenital cytomegalic inclusion disease. J. Pediatr. 1972, 81, 101–105. [Google Scholar] [CrossRef]
- Sacks, R.; Habermann, E. Pathological fracture in congenital rubella. A case report. JBJS 1977, 59, 557–559. [Google Scholar] [CrossRef]
- Smith, R.K.; Specht, E.E. Osseous lesions and pathologic fractures in congenital cytomegalic inclusion disease: Report of a case. Clin. Orthop. Relat. Res. 1979, 144, 280–283. [Google Scholar] [CrossRef]
- Li, Z.; Kong, K.; Qi, W. Osteoclast and its roles in calcium metabolism and bone development and remodeling. Biochem. Biophys. Res. Commun. 2006, 343, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Grigoriadis, A.E.; Wang, Z.-Q.; Cecchini, M.G.; Hofstetter, W.; Felix, R.; Fleisch, H.A.; Wagner, E.F. c-Fos: A Key Regulator of Osteoclast-Macrophage Lineage Determination and Bone Remodeling. Science 1994, 266, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Yao, Z.; Li, F.; Zhang, Q.; Badell, I.R.; Schwarz, E.M.; Takeshita, S.; Wagner, E.F.; Noda, M.; Matsuo, K.; et al. NF-kappaB p50 and p52 regulate receptor activator of NF-kappaB ligand (RANKL) and tumor necrosis factor-induced osteoclast precursor differentiation by activating c-Fos and NFATc1. J. Biol. Chem. 2007, 282, 18245–18253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, W.; Wang, X.; Yang, M.; Smith, L.C.; Dechow, P.C.; Sonoda, J.; Evans, R.M.; Wan, Y. PGC1beta mediates PPARgamma activation of osteoclastogenesis and rosiglitazone-induced bone loss. Cell. Metab. 2010, 11, 503–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Zhu, G.; Hao, L.; Wu, M.; Ci, H.; Li, Y.-P. C/EBPα regulates osteoclast lineage commitment. Proc. Natl. Acad. Sci. USA 2013, 110, 7294–7299. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.Q.; Ovitt, C.; Grigoriadis, A.E.; Möhle-Steinlein, U.; Rüther, U.; Wagner, E.F. Bone and haematopoietic defects in mice lacking c-fos. Nature 1992, 360, 741–745. [Google Scholar] [CrossRef]
- Mak, W.; Shao, X.; Dunstan, C.R.; Seibel, M.J.; Zhou, H. Biphasic Glucocorticoid-Dependent Regulation of Wnt Expression and Its Inhibitors in Mature Osteoblastic Cells. Calcif. Tissue Res. 2009, 85, 538–545. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.-P.; Paulson, C.; Shao, J.-Z.; Zhang, X.; Wu, M.; Chen, W. Wnt and the Wnt signaling pathway in bone development and disease. Front. Biosci. 2014, 19, 379–407. [Google Scholar] [CrossRef] [Green Version]
- Ponzetti, M.; Rucci, N. Updates on Osteoimmunology: What’s New on the Cross-Talk Between Bone and Immune System. Front. Endocrinol. 2019, 10, 236. [Google Scholar] [CrossRef]
- Yoshida, H.; Hayashi, S.-I.; Kunisada, T.; Ogawa, M.; Nishikawa, S.; Okamura, H.; Sudo, T.; Shultz, L.D.; Nishikawa, S.-I. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature 1990, 345, 442–444. [Google Scholar] [CrossRef]
- Wiktor-Jedrzejczak, W.; Bartocci, A.; Ferrante, A.W., Jr.; Ahmed-Ansari, A.; Sell, K.W.; Pollard, J.W.; Stanley, E.R. Total absence of colony-stimulating factor 1 in the macrophage-deficient osteopetrotic (op/op) mouse. Proc. Natl. Acad. Sci. USA 1990, 87, 4828–4832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudo, O.; Sabokbar, A.; Pocock, A.; Itonaga, I.; Fujikawa, Y.; Athanasou, N. Interleukin-6 and interleukin-11 support human osteoclast formation by a RANKL-independent mechanism. Bone 2003, 32, 1–7. [Google Scholar] [CrossRef]
- Blanchard, F.; Duplomb, L.; Baud’Huin, M.; Brounais, B. The dual role of IL-6-type cytokines on bone remodeling and bone tumors. Cytokine Growth Factor Rev. 2009, 20, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.; Schultz, R. Effect of infection by bovine viral diarrhea virus (BVDV) in vitro on interleukin-1 activity of bovine monocytes. Veter. Immunol. Immunopathol. 1991, 29, 251–265. [Google Scholar] [CrossRef]
- Shen, X.; He, Z.; Li, H.; Yao, C.; Zhang, Y.; He, L.; Li, S.; Huang, J.; Guo, Z. Distinct Functional Patterns of Gene Promoter Hypomethylation and Hypermethylation in Cancer Genomes. PLoS ONE 2012, 7, e44822. [Google Scholar] [CrossRef]
- Okuneva, O.; Li, Z.; Körber, I.; Tegelberg, S.; Joensuu, T.; Tian, L.; Lehesjoki, A.-E. Brain inflammation is accompanied by peripheral inflammation in Cstb−/−mice, a model for progressive myoclonus epilepsy. J. Neuroinflamm. 2016, 13, 298. [Google Scholar] [CrossRef] [Green Version]
- Laitala-Leinonen, T.; Rinne, R.; Saukko, P.; Väänänen, H.K.; Rinne, A. Cystatin B as an intracellular modulator of bone resorption. Matrix Biol. 2006, 25, 149–157. [Google Scholar] [CrossRef]
- Manninen, O.; Puolakkainen, T.; Lehto, J.; Harittu, E.; Kallonen, A.; Peura, M.; Laitala-Leinonen, T.; Kopra, O.; Kiviranta, R.; Lehesjoki, A.-E. Impaired osteoclast homeostasis in the cystatin B-deficient mouse model of progressive myoclonus epilepsy. Bone Rep. 2015, 3, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Bossowska-Nowicka, M.; Mielcarska, M.B.; Romaniewicz, M.; Kaczmarek, M.M.; Gregorczyk-Zboroch, K.P.; Struzik, J.; Grodzik, M.; Gieryńska, M.M.; Toka, F.N.; Szulc-Dąbrowska, L. Ectromelia virus suppresses expression of cathepsins and cystatins in conventional dendritic cells to efficiently execute the replication process. BMC Microbiol. 2019, 19, 92. [Google Scholar] [CrossRef]
- Choi, S.-W.; Yeon, J.-T.; Park, K.-I.; Lee, C.H.; Youn, B.S.; Oh, J.; Lee, M.S. VapB as a regulator of osteoclastogenesis via modulation of PLCγ2-Ca2+-NFAT signaling. FEBS Lett. 2012, 586, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Silbernagel, N.; Walecki, M.; Schäfer, M.K.-H.; Kessler, M.; Zobeiri, M.; Rinné, S.; Kiper, A.K.; Komadowski, M.A.; Vowinkel, K.S.; Wemhöner, K.; et al. The VAMP-associated protein VAPB is required for cardiac and neuronal pacemaker channel function. FASEB J. 2018, 32, 6159–6173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Piero, F.; Wilkins, P.A.; Dubovi, E.J. BVD virus antigen in Purkinje fibres in a calf. Veter. Rec. 1997, 140, 407–408. [Google Scholar]
- Yang, X.; Chen, D.; Long, H.; Zhu, B. The mechanisms of pathological extramedullary hematopoiesis in diseases. Cell. Mol. Life Sci. 2020, 77, 2723–2738. [Google Scholar] [CrossRef] [PubMed]
- Porter, B.F.; Ridpath, J.F.; Calise, D.V.; Payne, H.R.; Janke, J.J.; Baxter, D.G.; Edwards, J.F. Hypomyelination Associated with Bovine Viral Diarrhea Virus Type 2 Infection in a Longhorn Calf. Veter. Pathol. 2010, 47, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.; Hewicker, M.; Trautwein, G.; Pohlenz, J.; Liess, B. Viral Antigen Distribution in the Central Nervous System of Cattle Persistently Infected with Bovine Viral Diarrhea Virus. Veter. Pathol. 1989, 26, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Hewicker-Trautwein, M.; Liess, B.; Trautwein, G. Brain Lesions in Calves following Transplacental Infection with Bovine-virus Diarrhoea Virus. J. Veter. Med. Ser. B 1995, 42, 65–77. [Google Scholar] [CrossRef]
- Joensuu, T.; Tegelberg, S.; Reinmaa, E.; Segerstråle, M.; Hakala, P.; Pehkonen, H.; Korpi, E.R.; Tyynelä, J.; Taira, T.; Hovatta, I.; et al. Gene Expression Alterations in the Cerebellum and Granule Neurons of Cstb−/− Mouse Are Associated with Early Synaptic Changes and Inflammation. PLoS ONE 2014, 9, e89321. [Google Scholar] [CrossRef] [Green Version]
- Kabashi, E.; El Oussini, H.; Bercier, V.; Gros-Louis, F.; Valdmanis, P.; McDearmid, J.; Mejier, I.A.; Dion, P.A.; Dupre, N.; Hollinger, D.; et al. Investigating the contribution of VAPB/ALS8 loss of function in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2013, 22, 2350–2360. [Google Scholar] [CrossRef] [Green Version]
- Anagnostou, G.; Akbar, M.T.; Paul, P.; Angelinetta, C.; Steiner, T.J.; de Belleroche, J. Vesicle associated membrane protein B (VAPB) is decreased in ALS spinal cord. Neurobiol. Aging 2010, 31, 969–985. [Google Scholar] [CrossRef]
- Teuling, E.; Ahmed, S.; Haasdijk, E.; Demmers, J.; Steinmetz, M.; Akhmanova, A.; Jaarsma, D.; Hoogenraad, C.C. Motor Neuron Disease-Associated Mutant Vesicle-Associated Membrane Protein-Associated Protein (VAP) B Recruits Wild-Type VAPs into Endoplasmic Reticulum-Derived Tubular Aggregates. J. Neurosci. 2007, 27, 9801–9815. [Google Scholar] [CrossRef] [Green Version]
- Nosten-Bertrand, M.; Errington, M.L.; Murphy, K.P.S.J.; Tokugawa, Y.; Barboni, E.; Kozlova, E.; Michalovich, D.; Morris, R.G.M.; Silver, J.; Stewart, C.L.; et al. Normal spatial learning despite regional inhibition of LTP in mice lacking Thy-1. Nature 1996, 379, 826–829. [Google Scholar] [CrossRef] [PubMed]
- Mayeux-Portas, V.; File, S.E.; Stewart, C.L.; Morris, R.J. Mice lacking the cell adhesion molecule Thy-1 fail to use socially transmitted cues to direct their choice of food. Curr. Biol. 2000, 10, 68–75. [Google Scholar] [CrossRef] [Green Version]
- Bradley, J.E.; Ramirez, G.; Hagood, J.S. Roles and regulation of Thy-1, a context-dependent modulator of cell phenotype. BioFactors 2009, 35, 258–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, W.; Yang, X.Y.; Yang, T.T.; Factor, S.M.; Lisanti, M.P.; Molkentin, J.; Rincón, M.; Chow, C.-W. Requirement of transcription factor NFAT in developing atrial myocardium. J. Cell Biol. 2003, 161, 861–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graef, I.A.; Chen, F.; Crabtree, G.R. NFAT signaling in vertebrate development. Curr. Opin. Genet. Dev. 2001, 11, 505–512. [Google Scholar] [CrossRef]
- Kirkbride, C.A. Viral Agents and Associated Lesions Detected in a 10-Year Study of Bovine Abortions and Stillbirths. J. Veter. Diagn. Investig. 1992, 4, 374–379. [Google Scholar] [CrossRef] [Green Version]
- Breshears, M.A.; Johnson, B.J. Systemic Reactive Angioendotheliomatosis-like Syndrome in a Steer Presumed to be Persistently Infected with Bovine Viral Diarrhea Virus. Veter. Pathol. 2008, 45, 645–649. [Google Scholar] [CrossRef]
- Ghobadi, M.Z.; Mozhgani, S.-H.; Farzanehpour, M.; Behzadian, F. Identifying novel biomarkers of the pediatric influenza infection by weighted co-expression network analysis. Virol. J. 2019, 16, 124. [Google Scholar] [CrossRef] [Green Version]
- Lubick, K.J.; Robertson, S.J.; McNally, K.L.; Freedman, B.A.; Rasmussen, A.; Taylor, R.T.; Walts, A.D.; Tsuruda, S.; Sakai, M.; Ishizuka, M.; et al. Flavivirus Antagonism of Type I Interferon Signaling Reveals Prolidase as a Regulator of IFNAR1 Surface Expression. Cell Host Microbe 2015, 18, 61–74. [Google Scholar] [CrossRef] [Green Version]
- Riera, J.; Rodríguez, R.; Carcedo, M.T.; Campa, V.M.; Ramos, S.; Lazo, P.S. Isolation and characterization ofnudCfrom mouse macrophages, a gene implicated in the inflammatory response through the regulation of PAF-AH(I) activity. FEBS Lett. 2007, 581, 3057–3062. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.A.; Zhang, M.-Y.; Gocke, C.D.; De Souza, C.; Osmani, A.H.; Lynch, C.; Davies, J.; Bell, L.; Osmani, S.A. A homolog of the fungal nuclear migration gene nudC is involved in normal and malignant human hematopoiesis. Exp. Hematol. 1999, 27, 742–750. [Google Scholar] [CrossRef]
- Baigent, S.J.; Zhang, G.; Fray, M.D.; Flick-Smith, H.; Goodbourn, S.; McCauley, J.W. Inhibition of Beta Interferon Transcription by Noncytopathogenic Bovine Viral Diarrhea Virus Is through an Interferon Regulatory Factor 3-Dependent Mechanism. J. Virol. 2002, 76, 8979–8988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweizer, M.; Mätzener, P.; Pfaffen, G.; Stalder, H.; Peterhans, E. “Self” and “nonself” manipulation of interferon defense during persistent infection: Bovine viral diarrhea virus resists alpha/beta interferon without blocking antiviral activity against unrelated viruses replicating in its host cells. J. Virol. 2006, 80, 6926. [Google Scholar] [CrossRef] [Green Version]
- Hilton, L.; Moganeradj, K.; Zhang, G.; Chen, Y.-H.; Randall, R.E.; McCauley, J.W.; Goodbourn, S. The NPro Product of Bovine Viral Diarrhea Virus Inhibits DNA Binding by Interferon Regulatory Factor 3 and Targets It for Proteasomal Degradation. J. Virol. 2006, 80, 11723–11732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magkouras, I.; Mätzener, P.; Rümenapf, T.; Peterhans, E.; Schweizer, M. RNase-dependent inhibition of extracellular, but not intracellular, dsRNA-induced interferon synthesis by Erns of pestiviruses. J. Gen. Virol. 2008, 89, 2501–2506. [Google Scholar] [CrossRef] [PubMed]
- Darweesh, M.F.; Rajput, M.K.; Braun, L.J.; Rohila, J.S.; Chase, C.C. BVDV Npro protein mediates the BVDV induced immunosuppression through interaction with cellular S100A9 protein. Microb. Pathog. 2018, 121, 341–349. [Google Scholar] [CrossRef]
- Zhao, B.; Ivashkiv, L.B. Negative regulation of osteoclastogenesis and bone resorption by cytokines and transcriptional repressors. Arthritis Res. Ther. 2011, 13, 1–10, 234. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, T.; Shimakawa, G.; Tamura, M.; Yokosawa, H.; Arata, Y. ISG15 Regulates RANKL-Induced Osteoclastogenic Differentiation of RAW264 Cells. Biol. Pharm. Bull. 2015, 38, 482–486. [Google Scholar] [CrossRef] [Green Version]
- Driggers, R.W.; Ho, C.-Y.; Korhonen, E.M.; Kuivanen, S.; Jääskeläinen, A.J.; Smura, T.; Rosenberg, A.; Hill, D.A.; DeBiasi, R.L.; Vezina, G. Zika virus infection with prolonged maternal viremia and fetal brain abnormalities. N. Engl. J. Med. 2016, 374, 2142–2151. [Google Scholar] [CrossRef]
- Lee, S.H.; Shin, J.H.; Choi, B.M.; Kim, Y.-K. A Case of Cytomegalovirus Infection in a Neonate with Osteopetrosis. Pediatr. Infect. Vaccine 2016, 23, 72–76. [Google Scholar] [CrossRef]
- Katsafiloudi, M.; Gombakis, N.; Hatzipantelis, E.; Tragiannidis, A. Osteopetrorickets in an infant with coexistent congenital cytomegalovirus infection. Balk. J. Med Genet. 2020, 23, 107–110. [Google Scholar] [CrossRef]
- Ye, Z.; Wang, L.; Yang, T.; Chen, L.; Wang, T.; Chen, L.; Zhao, L.; Zhang, S.; Zheng, Z.; Luo, L.; et al. Maternal Viral Infection and Risk of Fetal Congenital Heart Diseases: A Meta-Analysis of Observational Studies. J. Am. Heart Assoc. 2019, 8, e011264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, L.K.; Velten, M. Maternal inflammation, growth retardation, and preterm birth: Insights into adult cardiovascular disease. Life Sci. 2011, 89, 417–421. [Google Scholar] [CrossRef]
- Barker, D.J.P.; Osmond, C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet 1986, 327, 1077–1081. [Google Scholar] [CrossRef]
- Lewis, A.J.; Austin, E.; Knapp, R.; Vaiano, T.; Galbally, M. Perinatal Maternal Mental Health, Fetal Programming and Child Development. Healthcare 2015, 3, 1212–1227. [Google Scholar] [CrossRef] [Green Version]
- Riaz, N.; Wolden, S.L.; Gelblum, D.Y.; Eric, J. Microbial Vertical Transmission during Human Pregnancy. Cell Host Microbe 2017, 21, 561–597. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Georges, H.M.; Van Campen, H.; Bielefeldt-Ohmann, H.; Hansen, T.R. Epigenomic and Proteomic Changes in Fetal Spleens Persistently Infected with Bovine Viral Diarrhea Virus: Repercussions for the Developing Immune System, Bone, Brain, and Heart. Viruses 2022, 14, 506. https://doi.org/10.3390/v14030506
Georges HM, Van Campen H, Bielefeldt-Ohmann H, Hansen TR. Epigenomic and Proteomic Changes in Fetal Spleens Persistently Infected with Bovine Viral Diarrhea Virus: Repercussions for the Developing Immune System, Bone, Brain, and Heart. Viruses. 2022; 14(3):506. https://doi.org/10.3390/v14030506
Chicago/Turabian StyleGeorges, Hanah M., Hana Van Campen, Helle Bielefeldt-Ohmann, and Thomas R. Hansen. 2022. "Epigenomic and Proteomic Changes in Fetal Spleens Persistently Infected with Bovine Viral Diarrhea Virus: Repercussions for the Developing Immune System, Bone, Brain, and Heart" Viruses 14, no. 3: 506. https://doi.org/10.3390/v14030506
APA StyleGeorges, H. M., Van Campen, H., Bielefeldt-Ohmann, H., & Hansen, T. R. (2022). Epigenomic and Proteomic Changes in Fetal Spleens Persistently Infected with Bovine Viral Diarrhea Virus: Repercussions for the Developing Immune System, Bone, Brain, and Heart. Viruses, 14(3), 506. https://doi.org/10.3390/v14030506