
Escape and Over-Activation of Innate Immune Responses by SARS-CoV-2: Two Faces of a Coin


Abstract
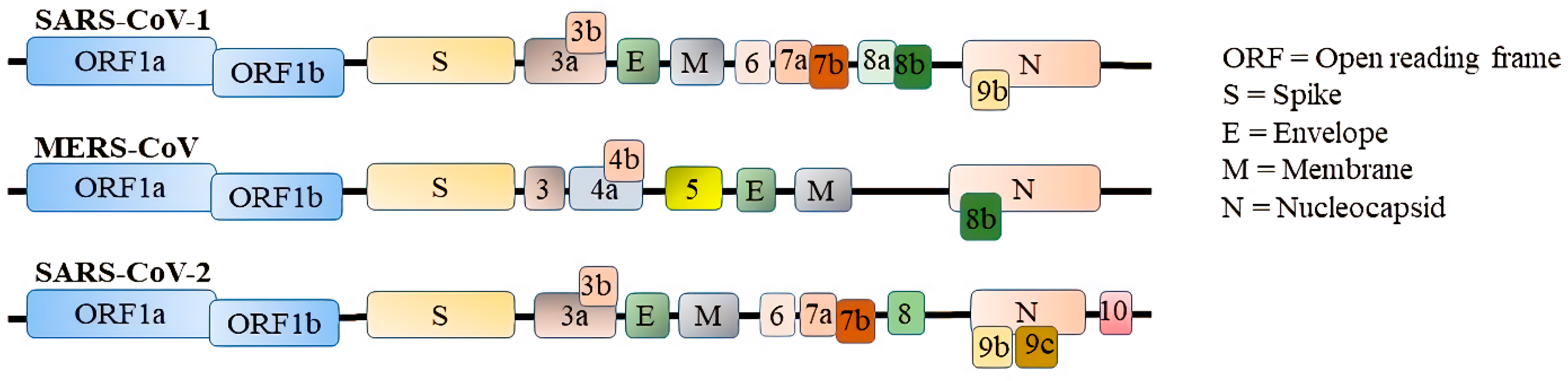
:1. Introduction
2. Host Innate Immune Sensors
3. Innate Immune Escape by CoVs
3.1. Sheltering and Modifying Viral RNA to Evade PRR Recognition
3.2. Inhibiting Host Protein Synthesis and the Degradation of Proteins
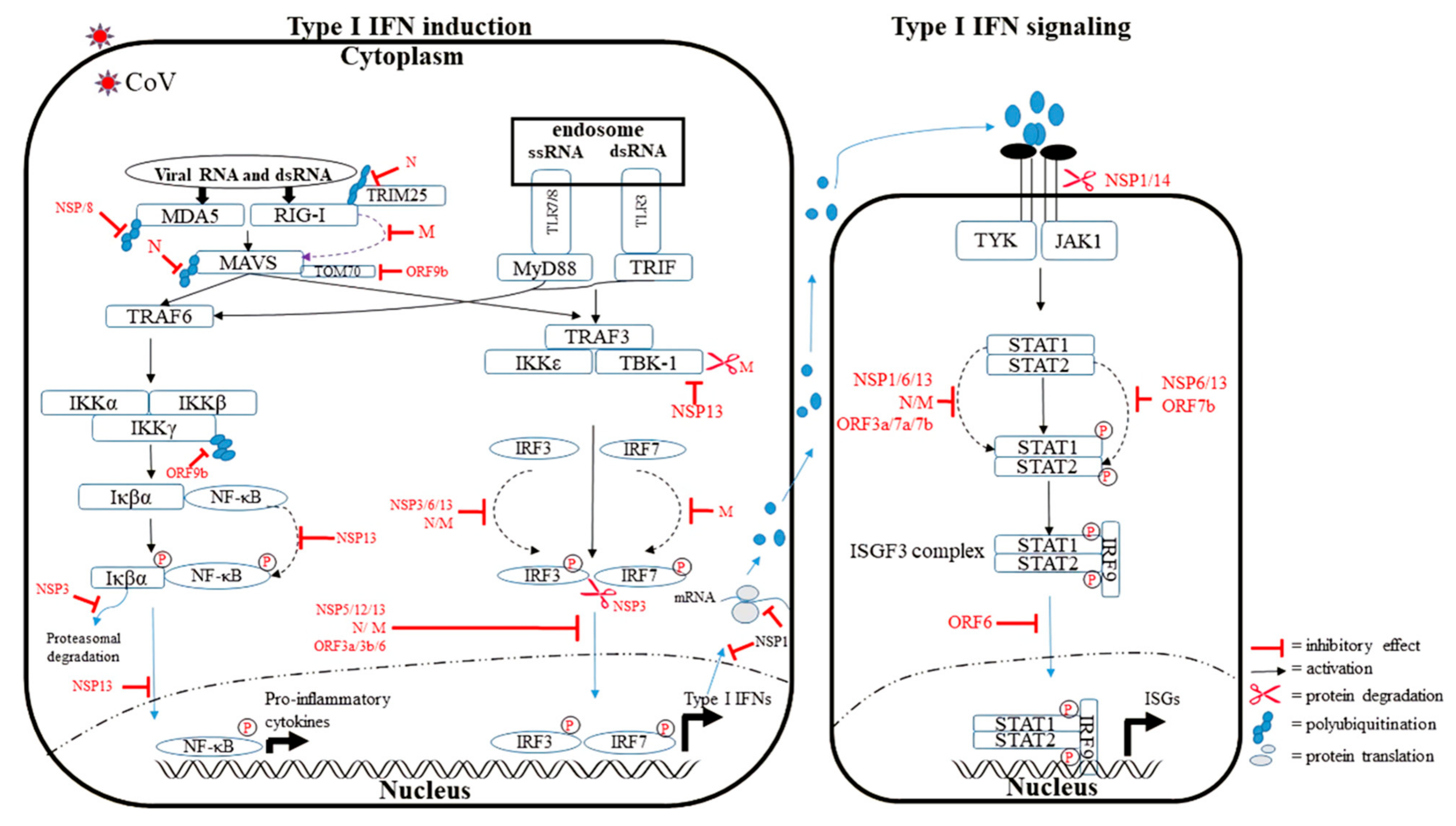
3.3. Impairing the Type I IFN Synthesis and Signaling Pathway
3.3.1. NSPs
NSP1
NSP3 and NSP6
NSP7, NSP8, and NSP12
NSP13
NSP14
3.3.2. Structural Proteins
N Protein
M Protein
3.3.3. Accessory Proteins
ORF3a
ORF3b
ORF6
ORF7
ORF9b
3.4. Innate Immune Evasion by Other CoV Viral Components
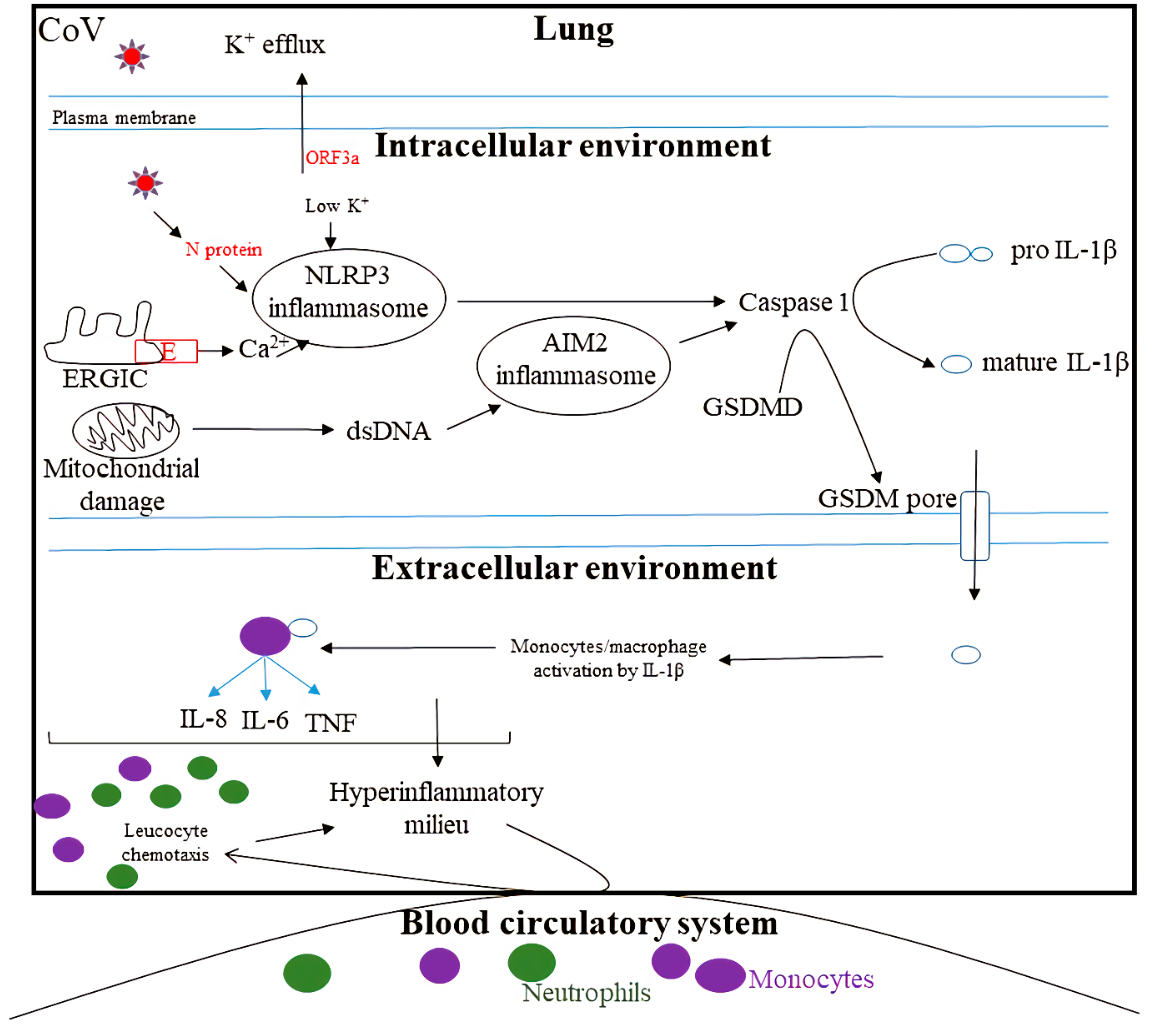
4. Inflammasome Activation by CoV
5. Therapeutics Targeting Innate Immune System
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, Z.; Qiu, Y.; Ge, X. The taxonomy, host range and pathogenicity of coronaviruses and other viruses in the Nidovirales order. Anim. Dis. 2021, 1, 5. [Google Scholar] [CrossRef] [PubMed]
- Pillaiyar, T.; Wendt, L.L.; Manickam, M.; Easwaran, M. The recent outbreaks of human coronaviruses: A medicinal chemistry perspective. Med. Res. Rev. 2021, 41, 72–135. [Google Scholar] [CrossRef] [PubMed]
- International Committee on Taxonomy of Viruses Executive Committee. The new scope of virus taxonomy: Partitioning the virosphere into 15 hierarchical ranks. Nat. Microbiol. 2020, 5, 668. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, T.S.; de Sá, K.S.; Ishimoto, A.Y.; Becerra, A.; Oliveira, S.; Almeida, L.; Gonçalves, A.V.; Perucello, D.B.; Andrade, W.A.; Castro, R. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J. Exp. Med. 2021, 218, e20201707. [Google Scholar] [CrossRef]
- Junqueira, C.; Crespo, Â.; Ranjbar, S.; Lewandrowski, M.; Ingber, J.; de Lacerda, L.B.; Parry, B.; Ravid, S.; Clark, S.; Ho, F. SARS-CoV-2 infects blood monocytes to activate NLRP3 and AIM2 inflammasomes, pyroptosis and cytokine release. Res. Sq. 2021. [Google Scholar] [CrossRef]
- Satış, H.; Özger, H.S.; Yıldız, P.A.; Hızel, K.; Gulbahar, Ö.; Erbaş, G.; Aygencel, G.; Tunccan, O.G.; Öztürk, M.A.; Dizbay, M. Prognostic value of interleukin-18 and its association with other inflammatory markers and disease severity in COVID-19. Cytokine 2021, 137, 155302. [Google Scholar] [CrossRef]
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020, 584, 463–469. [Google Scholar] [CrossRef]
- Ito, H.; Kimura, H.; Karasawa, T.; Hisata, S.; Sadatomo, A.; Inoue, Y.; Yamada, N.; Aizawa, E.; Hishida, E.; Kamata, R. NLRP3 inflammasome activation in lung vascular endothelial cells contributes to intestinal ischemia/reperfusion-induced acute lung injury. J. Immunol. 2020, 205, 1393–1405. [Google Scholar] [CrossRef]
- Loo, Y.-M.; Gale, M., Jr. Immune signaling by RIG-I-like receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reikine, S.; Nguyen, J.B.; Modis, Y. Pattern recognition and signaling mechanisms of RIG-I and MDA5. Front. Immunol. 2014, 5, 342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbas, A.K.; Lichtman, A.H.; Pillai, S. Cellular and Molecular Immunology E-Book; Elsevier Health Sciences: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Hou, F.; Sun, L.; Zheng, H.; Skaug, B.; Jiang, Q.-X.; Chen, Z.J. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 2011, 146, 448–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onomoto, K.; Onoguchi, K.; Yoneyama, M. Regulation of RIG-I-like receptor-mediated signaling: Interaction between host and viral factors. Cell. Mol. Immunol. 2021, 18, 539–555. [Google Scholar] [CrossRef] [PubMed]
- Stark, G.R.; Darnell, J.E., Jr. The JAK-STAT pathway at twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef] [Green Version]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [Green Version]
- Balachandran, S.; Roberts, P.C.; Brown, L.E.; Truong, H.; Pattnaik, A.K.; Archer, D.R.; Barber, G.N. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity 2000, 13, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. Correction: Corrigendum: A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2015, 525, 144. [Google Scholar]
- Platanias, L.C. Mechanisms of type-I-and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Kouwaki, T.; Nishimura, T.; Wang, G.; Oshiumi, H. RIG-I-like receptor-mediated recognition of viral genomic RNA of severe acute respiratory syndrome coronavirus-2 and viral escape from the host innate immune responses. Front. Immunol. 2021, 12, 700926. [Google Scholar] [CrossRef]
- Rebendenne, A.; Chaves Valadão, A.L.; Tauziet, M.; Maarifi, G.; Bonaventure, B.; McKellar, J.; Planès, R.; Nisole, S.; Arnaud-Arnould, M.; Moncorgé, O. SARS-CoV-2 triggers an MDA-5-dependent interferon response which is unable to control replication in lung epithelial cells. J. Virol. 2021, 95, e02415–e02420. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, N.G.; Chauveau, L.; Hertzog, J.; Bridgeman, A.; Fowler, G.; Moonen, J.P.; Dupont, M.; Russell, R.A.; Noerenberg, M.; Rehwinkel, J. The RNA sensor MDA5 detects SARS-CoV-2 infection. Sci. Rep. 2021, 11, 13638. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Riva, L.; Pu, Y.; Martin-Sancho, L.; Kanamune, J.; Yamamoto, Y.; Sakai, K.; Gotoh, S.; Miorin, L.; De Jesus, P.D. MDA5 governs the innate immune response to SARS-CoV-2 in lung epithelial cells. Cell Rep. 2021, 34, 108628. [Google Scholar] [CrossRef]
- Wu, J.; Shi, Y.; Pan, X.; Wu, S.; Hou, R.; Zhang, Y.; Zhong, T.; Tang, H.; Du, W.; Wang, L. SARS-CoV-2 ORF9b inhibits RIG-I-MAVS antiviral signaling by interrupting K63-linked ubiquitination of NEMO. Cell Rep. 2021, 34, 108761. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Lee, J.-H.; Parker, Z.M.; Acharya, D.; Chiang, J.J.; van Gent, M.; Riedl, W.; Davis-Gardner, M.E.; Wies, E.; Chiang, C. ISG15-dependent activation of the sensor MDA5 is antagonized by the SARS-CoV-2 papain-like protease to evade host innate immunity. Nat. Microbiol. 2021, 6, 467–478. [Google Scholar] [CrossRef]
- Durand, J.K.; Zhang, Q.; Baldwin, A.S. Roles for the IKK-related kinases TBK1 and IKKε in cancer. Cells 2018, 7, 139. [Google Scholar] [CrossRef] [Green Version]
- Zheng, M.; Karki, R.; Williams, E.P.; Yang, D.; Fitzpatrick, E.; Vogel, P.; Jonsson, C.B.; Kanneganti, T.-D. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 2021, 22, 829–838. [Google Scholar] [CrossRef]
- Zhao, Y.; Kuang, M.; Li, J.; Zhu, L.; Jia, Z.; Guo, X.; Hu, Y.; Kong, J.; Yin, H.; Wang, X. SARS-CoV-2 spike protein interacts with and activates TLR41. Cell Res. 2021, 31, 818–820. [Google Scholar] [CrossRef]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570b. [Google Scholar] [CrossRef]
- Van Der Made, C.I.; Simons, A.; Schuurs-Hoeijmakers, J.; Van Den Heuvel, G.; Mantere, T.; Kersten, S.; Van Deuren, R.C.; Steehouwer, M.; Van Reijmersdal, S.V.; Jaeger, M. Presence of genetic variants among young men with severe COVID-19. JAMA 2020, 324, 663–673. [Google Scholar] [CrossRef]
- Asano, T.; Boisson, B.; Onodi, F.; Matuozzo, D.; Moncada-Velez, M.; Maglorius Renkilaraj, M.R.L.; Zhang, P.; Meertens, L.; Bolze, A.; Materna, M.; et al. X-linked recessive TLR7 deficiency in ~1% of men under 60 years old with life-threatening COVID-19. Sci. Immunol. 2021, 6, eabl4348. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- He, W.-t.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.-H.; Zhong, C.-Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Zhao, C.; Zhao, W. NLRP3 Inflammasome—A Key Player in Antiviral Responses. Front. Immunol. 2020, 11, 211. [Google Scholar] [CrossRef] [Green Version]
- Freeman, T.L.; Swartz, T.H. Targeting the NLRP3 inflammasome in severe COVID-19. Front. Immunol. 2020, 11, 1518. [Google Scholar] [CrossRef]
- Lee, J.Y.; Bae, S.; Myoung, J. Middle East respiratory syndrome coronavirus-encoded accessory proteins impair MDA5-and TBK1-mediated activation of NF-κB. J. Microbiol. Biotechnol. 2019, 29, 1316–1323. [Google Scholar] [CrossRef]
- Nieto-Torres, J.L.; Verdiá-Báguena, C.; Jimenez-Guardeño, J.M.; Regla-Nava, J.A.; Castaño-Rodriguez, C.; Fernandez-Delgado, R.; Torres, J.; Aguilella, V.M.; Enjuanes, L. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology 2015, 485, 330–339. [Google Scholar] [CrossRef] [Green Version]
- Chen, I.-Y.; Moriyama, M.; Chang, M.-F.; Ichinohe, T. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front. Microbiol. 2019, 10, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siu, K.L.; Yuen, K.S.; Castano-Rodriguez, C.; Ye, Z.W.; Yeung, M.L.; Fung, S.Y.; Yuan, S.; Chan, C.P.; Yuen, K.Y.; Enjuanes, L. Severe acute respiratory syndrome Coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019, 33, 8865–8877. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-C.; Krüger, J.; Sramala, I.; Hsu, H.-J.; Henklein, P.; Chen, Y.-M.A.; Fischer, W.B. ORF8a of SARS-CoV forms an ion channel: Experiments and molecular dynamics simulations. Biochim. Biophys. Acta (BBA) Biomembr. 2011, 1808, 572–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, C.-S.; Nabar, N.R.; Huang, N.-N.; Kehrl, J.H. SARS-Coronavirus Open Reading Frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 2019, 5, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayn, M.; Hirschenberger, M.; Koepke, L.; Nchioua, R.; Straub, J.H.; Klute, S.; Hunszinger, V.; Zech, F.; Bozzo, C.P.; Aftab, W. Systematic functional analysis of SARS-CoV-2 proteins uncovers viral innate immune antagonists and remaining vulnerabilities. Cell Rep. 2021, 35, 109126. [Google Scholar] [CrossRef]
- Hartenian, E.; Nandakumar, D.; Lari, A.; Ly, M.; Tucker, J.M.; Glaunsinger, B.A. The molecular virology of coronaviruses. J. Biol. Chem. 2020, 295, 12910–12934. [Google Scholar] [CrossRef]
- Hagemeijer, M.C.; Monastyrska, I.; Griffith, J.; van der Sluijs, P.; Voortman, J.; en Henegouwen, P.M.v.B.; Vonk, A.M.; Rottier, P.J.; Reggiori, F.; De Haan, C.A. Membrane rearrangements mediated by coronavirus nonstructural proteins 3 and 4. Virology 2014, 458, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Decroly, E.; Ferron, F.; Lescar, J.; Canard, B. Conventional and unconventional mechanisms for capping viral mRNA. Nat. Rev. Microbiol. 2012, 10, 51–65. [Google Scholar] [CrossRef]
- Bouvet, M.; Debarnot, C.; Imbert, I.; Selisko, B.; Snijder, E.J.; Canard, B.; Decroly, E. In vitro reconstitution of SARS-coronavirus mRNA cap methylation. PLoS Pathog. 2010, 6, e1000863. [Google Scholar] [CrossRef]
- Chen, Y.; Su, C.; Ke, M.; Jin, X.; Xu, L.; Zhang, Z.; Wu, A.; Sun, Y.; Yang, Z.; Tien, P. Biochemical and structural insights into the mechanisms of SARS coronavirus RNA ribose 2′-O-methylation by nsp16/nsp10 protein complex. PLoS Pathog. 2011, 7, e1002294. [Google Scholar] [CrossRef] [Green Version]
- Menachery, V.D.; Debbink, K.; Baric, R.S. Coronavirus non-structural protein 16: Evasion, attenuation, and possible treatments. Virus Res. 2014, 194, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Züst, R.; Cervantes-Barragan, L.; Habjan, M.; Maier, R.; Neuman, B.W.; Ziebuhr, J.; Szretter, K.J.; Baker, S.C.; Barchet, W.; Diamond, M.S. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 2011, 12, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilamowski, M.; Sherrell, D.A.; Minasov, G.; Kim, Y.; Shuvalova, L.; Lavens, A.; Chard, R.; Maltseva, N.; Jedrzejczak, R.; Rosas-Lemus, M. 2′-O methylation of RNA cap in SARS-CoV-2 captured by serial crystallography. Proc. Natl. Acad. Sci. USA 2021, 118, e2100170118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Miorin, L.; Makio, T.; Dehghan, I.; Gao, S.; Xie, Y.; Zhong, H.; Esparza, M.; Kehrer, T.; Kumar, A. Nsp1 protein of SARS-CoV-2 disrupts the mRNA export machinery to inhibit host gene expression. Sci. Adv. 2021, 7, eabe7386. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Ishida, R.; Strilets, T.; Cole, J.; Lopez-Orozco, J.; Fayad, N.; Felix-Lopez, A.; Elaish, M.; Evseev, D.; Magor, K.E. SARS-CoV-2 non-structural protein 1 inhibits the interferon response by causing depletion of key host signaling factors. J. Virol. 2021, 95, e00266-21. [Google Scholar] [CrossRef]
- Narayanan, K.; Huang, C.; Lokugamage, K.; Kamitani, W.; Ikegami, T.; Tseng, C.-T.K.; Makino, S. Severe acute respiratory syndrome coronavirus nsp1 suppresses host gene expression, including that of type I interferon, in infected cells. J. Virol. 2008, 82, 4471–4479. [Google Scholar] [CrossRef] [Green Version]
- Hsu, J.C.-C.; Laurent-Rolle, M.; Pawlak, J.B.; Wilen, C.B.; Cresswell, P. Translational shutdown and evasion of the innate immune response by SARS-CoV-2 NSP14 protein. Proc. Natl. Acad. Sci. USA 2021, 118, e2101161118. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wu, L.; Shaw, N.; Gao, Y.; Wang, J.; Sun, Y.; Lou, Z.; Yan, L.; Zhang, R.; Rao, Z. Structural basis and functional analysis of the SARS coronavirus nsp14–nsp10 complex. Proc. Natl. Acad. Sci. USA 2015, 112, 9436–9441. [Google Scholar] [CrossRef] [Green Version]
- Bouvet, M.; Lugari, A.; Posthuma, C.C.; Zevenhoven, J.C.; Bernard, S.; Betzi, S.; Imbert, I.; Canard, B.; Guillemot, J.-C.; Lécine, P. Coronavirus Nsp10, a critical co-factor for activation of multiple replicative enzymes. J. Biol. Chem. 2014, 289, 25783–25796. [Google Scholar]
- Banerjee, A.K.; Blanco, M.R.; Bruce, E.A.; Honson, D.D.; Chen, L.M.; Chow, A.; Bhat, P.; Ollikainen, N.; Quinodoz, S.A.; Loney, C. SARS-CoV-2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell 2020, 183, 1325–1339.e1321. [Google Scholar] [CrossRef]
- Sui, L.; Zhao, Y.; Wang, W.; Wu, P.; Wang, Z.; Yu, Y.; Hou, Z.; Tan, G.; Liu, Q. SARS-CoV-2 membrane protein inhibits type I interferon production through ubiquitin-mediated degradation of TBK1. Front. Immunol. 2021, 12, 662989. [Google Scholar] [CrossRef]
- Moustaqil, M.; Ollivier, E.; Chiu, H.-P.; Van Tol, S.; Rudolffi-Soto, P.; Stevens, C.; Bhumkar, A.; Hunter, D.J.; Freiberg, A.N.; Jacques, D. SARS-CoV-2 proteases PLpro and 3CLpro cleave IRF3 and critical modulators of inflammatory pathways (NLRP12 and TAB1): Implications for disease presentation across species. Emerg. Microb. Infect. 2021, 10, 178–195. [Google Scholar] [CrossRef]
- Shi, C.-S.; Qi, H.-Y.; Boularan, C.; Huang, N.-N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyts, I.; Bucciol, G.; Quinti, I.; Neven, B.; Fischer, A.; Seoane, E.; Lopez-Granados, E.; Gianelli, C.; Robles-Marhuenda, A.; Jeandel, P.Y.; et al. Coronavirus disease 2019 in patients with inborn errors of immunity: An international study. J. Allergy Clin. Immunol. 2021, 147, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Goudouris, E.S.; Pinto-Mariz, F.; Mendonça, L.O.; Aranda, C.S.; Guimarães, R.R.; Kokron, C.; Barros, M.T.; Anísio, F.; Alonso, M.L.O.; Marcelino, F.; et al. Outcome of SARS-CoV-2 Infection in 121 Patients with Inborn Errors of Immunity: A Cross-Sectional Study. J. Clin. Immunol. 2021, 41, 1479–1489. [Google Scholar] [CrossRef]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.-H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Béziat, V. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Cao, Z.; Xie, X.; Zhang, X.; Chen, J.Y.-C.; Wang, H.; Menachery, V.D.; Rajsbaum, R.; Shi, P.-Y. Evasion of type I interferon by SARS-CoV-2. Cell Rep. 2020, 33, 108234. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, S.G.; Wang, N.; Chen, Z.; Chen, Z.; Tseng, M.; Barretto, N.; Lin, R.; Peters, C.J.; Tseng, C.-T.K.; Baker, S.C. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J. Biol. Chem. 2007, 282, 32208–32221. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Yin, W.; Xu, H.E. RNA-dependent RNA polymerase: Structure, mechanism, and drug discovery for COVID-19. Biochem. Biophys. Res. Commun. 2021, 538, 47–53. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, X.; Wang, F.; Wang, P.; Li, X.; Kuang, E. Suppression of MDA5-mediated antiviral immune responses by NSP8 of SARS-CoV-2. bioRxiv 2020. [Google Scholar]
- Wang, W.; Zhou, Z.; Xiao, X.; Tian, Z.; Dong, X.; Wang, C.; Li, L.; Ren, L.; Lei, X.; Xiang, Z. SARS-CoV-2 nsp12 attenuates type I interferon production by inhibiting IRF3 nuclear translocation. Cell. Mol. Immunol. 2021, 18, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Gao, M.; Gao, X.; Zhu, B.; Huang, J.; Luo, K.; Zhang, Y.; Sun, J.; Deng, M.; Lou, Z. SARS-CoV-2 non-structural protein 13 (nsp13) hijacks host deubiquitinase USP13 and counteracts host antiviral immune response. Signal Transduct. Target. Ther. 2021, 6, 119. [Google Scholar] [CrossRef]
- Lei, X.; Dong, X.; Ma, R.; Wang, W.; Xiao, X.; Tian, Z.; Wang, C.; Wang, Y.; Li, L.; Ren, L. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 2020, 11, 3810. [Google Scholar] [CrossRef]
- Yuen, C.-K.; Lam, J.-Y.; Wong, W.-M.; Mak, L.-F.; Wang, X.; Chu, H.; Cai, J.-P.; Jin, D.-Y.; To, K.K.-W.; Chan, J.F.-W. SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerg. Microb. Infect. 2020, 9, 1418–1428. [Google Scholar] [CrossRef]
- Hu, Y.; Li, W.; Gao, T.; Cui, Y.; Jin, Y.; Li, P.; Ma, Q.; Liu, X.; Cao, C. The severe acute respiratory syndrome coronavirus nucleocapsid inhibits type I interferon production by interfering with TRIM25-mediated RIG-I ubiquitination. J. Virol. 2017, 91, e02143-16. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Pan, J.a.; Tao, J.; Guo, D. SARS-CoV nucleocapsid protein antagonizes IFN-β response by targeting initial step of IFN-β induction pathway, and its C-terminal region is critical for the antagonism. Virus Genes 2011, 42, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Gori Savellini, G.; Anichini, G.; Gandolfo, C.; Cusi, M.G. SARS-CoV-2 N protein targets TRIM25-mediated RIG-I activation to suppress innate immunity. Viruses 2021, 13, 1439. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Dai, T.; Qin, Z.; Pan, T.; Chu, F.; Lou, L.; Zhang, L.; Yang, B.; Huang, H.; Lu, H. Targeting liquid–liquid phase separation of SARS-CoV-2 nucleocapsid protein promotes innate antiviral immunity by elevating MAVS activity. Nat. Cell Biol. 2021, 23, 718–732. [Google Scholar] [CrossRef] [PubMed]
- Mu, J.; Fang, Y.; Yang, Q.; Shu, T.; Wang, A.; Huang, M.; Jin, L.; Deng, F.; Qiu, Y.; Zhou, X. SARS-CoV-2 N protein antagonizes type I interferon signaling by suppressing phosphorylation and nuclear translocation of STAT1 and STAT2. Cell Discov. 2020, 6, 65. [Google Scholar] [CrossRef]
- Zhao, Y.; Sui, L.; Wu, P.; Wang, W.; Wang, Z.; Yu, Y.; Hou, Z.; Tan, G.; Liu, Q.; Wang, G. A dual-role of SARS-CoV-2 nucleocapsid protein in regulating innate immune response. Signal Transduct. Target. Ther. 2021, 6, 331. [Google Scholar] [CrossRef]
- Siu, K.-L.; Kok, K.-H.; Ng, M.-H.J.; Poon, V.K.; Yuen, K.-Y.; Zheng, B.-J.; Jin, D.-Y. Severe acute respiratory syndrome coronavirus M protein inhibits type I interferon production by impeding the formation of TRAF3·TANK·TBK1/IKKϵ complex. J. Biol. Chem. 2009, 284, 16202–16209. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Zhuang, M.-W.; Han, L.; Zhang, J.; Nan, M.-L.; Zhan, P.; Kang, D.; Liu, X.; Gao, C.; Wang, P.-H. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) membrane (M) protein inhibits type I and III interferon production by targeting RIG-I/MDA-5 signaling. Signal Transduct. Target. Ther. 2020, 5, 299. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.-Z.; Wang, S.-Y.; Zheng, Z.-Q.; Huang, Y.; Li, W.-W.; Xu, Z.-S.; Wang, Y.-Y. SARS-CoV-2 membrane glycoprotein M antagonizes the MAVS-mediated innate antiviral response. Cell. Mol. Immunol. 2021, 18, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Minakshi, R.; Padhan, K.; Rani, M.; Khan, N.; Ahmad, F.; Jameel, S. The SARS Coronavirus 3a protein causes endoplasmic reticulum stress and induces ligand-independent downregulation of the type 1 interferon receptor. PLoS ONE 2009, 4, e8342. [Google Scholar] [CrossRef] [PubMed]
- Konno, Y.; Kimura, I.; Uriu, K.; Fukushi, M.; Irie, T.; Koyanagi, Y.; Sauter, D.; Gifford, R.J.; Nakagawa, S.; Sato, K. SARS-CoV-2 ORF3b is a potent interferon antagonist whose activity is increased by a naturally occurring elongation variant. Cell Rep. 2020, 32, 108185. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Hon, C.-C.; Li, Y.; Wang, D.; Xu, G.; Zhang, H.; Zhou, P.; Poon, L.L.; Lam, T.T.-Y.; Leung, F.C.-C. Intraspecies diversity of SARS-like coronaviruses in Rhinolophus sinicus and its implications for the origin of SARS coronaviruses in humans. J. Gen. Virol. 2010, 91, 1058–1062. [Google Scholar] [CrossRef] [Green Version]
- Frieman, M.; Yount, B.; Heise, M.; Kopecky-Bromberg, S.A.; Palese, P.; Baric, R.S. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J. Virol. 2007, 81, 9812–9824. [Google Scholar] [CrossRef] [Green Version]
- Addetia, A.; Lieberman, N.A.; Phung, Q.; Hsiang, T.-Y.; Xie, H.; Roychoudhury, P.; Shrestha, L.; Loprieno, M.A.; Huang, M.-L.; Gale Jr, M. SARS-CoV-2 ORF6 Disrupts Bidirectional Nucleocytoplasmic Transport through Interactions with Rae1 and Nup98. Mbio 2021, 12, e00065-21. [Google Scholar] [CrossRef]
- Miorin, L.; Kehrer, T.; Sanchez-Aparicio, M.T.; Zhang, K.; Cohen, P.; Patel, R.S.; Cupic, A.; Makio, T.; Mei, M.; Moreno, E. SARS-CoV-2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 28344–28354. [Google Scholar] [CrossRef]
- Yoo, J.-S.; Sasaki, M.; Cho, S.X.; Kasuga, Y.; Zhu, B.; Ouda, R.; Orba, Y.; de Figueiredo, P.; Sawa, H.; Kobayashi, K.S. SARS-CoV-2 inhibits induction of the MHC class I pathway by targeting the STAT1-IRF1-NLRC5 axis. Nat. Commun. 2021, 12, 6602. [Google Scholar] [CrossRef]
- Mattoo, S.; Myoung, J. A Promising Vaccination Strategy against COVID-19 on the Horizon: Heterologous Immunization. J. Microbiol. Biotechnol. 2021, 31, 1601–1614. [Google Scholar] [CrossRef] [PubMed]
- Arya, R.; Kumari, S.; Pandey, B.; Mistry, H.; Bihani, S.C.; Das, A.; Prashar, V.; Gupta, G.D.; Panicker, L.; Kumar, M. Structural insights into SARS-CoV-2 proteins. J. Mol. Biol. 2021, 433, 166725. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.-W.; Zhang, H.-N.; Meng, Q.-F.; Xie, J.; Li, Y.; Chen, H.; Zheng, Y.-X.; Wang, X.-N.; Qi, H.; Zhang, J. SARS-CoV-2 Orf9b suppresses type I interferon responses by targeting TOM70. Cell. Mol. Immunol. 2020, 17, 998–1000. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Bae, S.; Myoung, J. Middle East respiratory syndrome coronavirus-encoded ORF8b strongly antagonizes IFN-β promoter activation: Its implication for vaccine design. J. Microbiol. 2019, 57, 803–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Akinyemi, I.A.; Chitre, S.A.; Loeb, J.C.; Lednicky, J.A.; McIntosh, M.T.; Bhaduri-McIntosh, S. SARS-CoV-2 viroporin encoded by ORF3a triggers the NLRP3 inflammatory pathway. Virology 2022, 568, 13–22. [Google Scholar] [CrossRef]
- Xia, B.; Shen, X.; He, Y.; Pan, X.; Liu, F.-L.; Wang, Y.; Yang, F.; Fang, S.; Wu, Y.; Duan, Z. SARS-CoV-2 envelope protein causes acute respiratory distress syndrome (ARDS)-like pathological damages and constitutes an antiviral target. Cell Res. 2021, 31, 847–860. [Google Scholar] [CrossRef]
- Pan, P.; Shen, M.; Yu, Z.; Ge, W.; Chen, K.; Tian, M.; Xiao, F.; Wang, Z.; Wang, J.; Jia, Y. SARS-CoV-2 N protein promotes NLRP3 inflammasome activation to induce hyperinflammation. Nat. Commun. 2021, 12, 4664. [Google Scholar] [CrossRef]
- Van de Veerdonk, F.L.; Netea, M.G. Blocking IL-1 to prevent respiratory failure in COVID-19. Crit. Care 2020, 24, 1–6. [Google Scholar] [CrossRef]
- Xiong, S.; Hong, Z.; Huang, L.S.; Tsukasaki, Y.; Nepal, S.; Di, A.; Zhong, M.; Wu, W.; Ye, Z.; Gao, X. IL-1β suppression of VE-cadherin transcription underlies sepsis-induced inflammatory lung injury. J. Clin. Investig. 2020, 130, 3684–3698. [Google Scholar] [CrossRef]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef]
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute respiratory distress syndrome. Nat. Rev. Dis. Primers 2019, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Costela-Ruiz, V.J.; Illescas-Montes, R.; Puerta-Puerta, J.M.; Ruiz, C.; Melguizo-Rodríguez, L. SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev. 2020, 54, 62–75. [Google Scholar] [CrossRef]
- Ma, J.; Zhu, F.; Zhao, M.; Shao, F.; Yu, D.; Ma, J.; Zhang, X.; Li, W.; Qian, Y.; Zhang, Y. SARS-CoV-2 nucleocapsid suppresses host pyroptosis by blocking Gasdermin D cleavage. EMBO J. 2021, 40, e108249. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.C.; Soares, V.C.; de Azevedo-Quintanilha, I.G.; Dias, S.d.S.G.; Fintelman-Rodrigues, N.; Sacramento, C.Q.; Mattos, M.; de Freitas, C.S.; Temerozo, J.R.; Teixeira, L. SARS-CoV-2 engages inflammasome and pyroptosis in human primary monocytes. Cell Death Discov. 2021, 7, 43. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.-E.; Kim, D.-K.; Song, Y.-J. SARS-CoV-2 nonstructural proteins 1 and 13 suppress caspase-1 and the NLRP3 inflammasome activation. Microorganisms 2021, 9, 494. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Emergency Use Authorization. Available online: https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization#coviddrugs (accessed on 28 February 2022).
- Hoagland, D.A.; Møller, R.; Uhl, S.A.; Oishi, K.; Frere, J.; Golynker, I.; Horiuchi, S.; Panis, M.; Blanco-Melo, D.; Sachs, D. Leveraging the antiviral type I interferon system as a first line of defense against SARS-CoV-2 pathogenicity. Immunity 2021, 54, 557–570.e555. [Google Scholar] [CrossRef]
- Wang, N.; Zhan, Y.; Zhu, L.; Hou, Z.; Liu, F.; Song, P.; Qiu, F.; Wang, X.; Zou, X.; Wan, D. Retrospective multicenter cohort study shows early interferon therapy is associated with favorable clinical responses in COVID-19 patients. Cell Host Microb. 2020, 28, 455–464. [Google Scholar] [CrossRef]
- Mao, T.; Israelow, B.; Lucas, C.; Vogels, C.B.; Gomez-Calvo, M.L.; Fedorova, O.; Breban, M.I.; Menasche, B.L.; Dong, H.; Linehan, M. A stem-loop RNA RIG-I agonist protects against acute and chronic SARS-CoV-2 infection in mice. J. Exp. Med. 2021, 219, e20211818. [Google Scholar] [CrossRef]
- Galani, I.E.; Triantafyllia, V.; Eleminiadou, E.-E.; Koltsida, O.; Stavropoulos, A.; Manioudaki, M.; Thanos, D.; Doyle, S.E.; Kotenko, S.V.; Thanopoulou, K. Interferon-λ mediates non-redundant front-line antiviral protection against influenza virus infection without compromising host fitness. Immunity 2017, 46, 875–890.e876. [Google Scholar] [CrossRef]
- Felgenhauer, U.; Schoen, A.; Gad, H.H.; Hartmann, R.; Schaubmar, A.R.; Failing, K.; Drosten, C.; Weber, F. Inhibition of SARS–CoV-2 by type I and type III interferons. J. Biol. Chem. 2020, 295, 13958–13964. [Google Scholar] [CrossRef]
- Stanifer, M.L.; Kee, C.; Cortese, M.; Zumaran, C.M.; Triana, S.; Mukenhirn, M.; Kraeusslich, H.-G.; Alexandrov, T.; Bartenschlager, R.; Boulant, S. Critical role of type III interferon in controlling SARS-CoV-2 infection in human intestinal epithelial cells. Cell Rep. 2020, 32, 107863. [Google Scholar] [CrossRef] [PubMed]
- Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Edwards, C.E.; Martinez, D.R.; Montgomery, S.A.; West, A.; Yount, B.L.; Hou, Y.J.; Adams, L.E. A mouse-adapted model of SARS-CoV-2 to test COVID-19 countermeasures. Nature 2020, 586, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Shahbazi, M.; Amri Maleh, P.; Bagherzadeh, M.; Moulana, Z.; Sepidarkish, M.; Rezanejad, M.; Mirzakhani, M.; Ebrahimpour, S.; Ghorbani, H.; Ahmadnia, Z. Linkage of Lambda Interferons in Protection Against Severe COVID-19. J. Interferon Cytokine Res. 2021, 41, 149–152. [Google Scholar] [CrossRef]
- Fukuda, Y.; Homma, T.; Inoue, H.; Onitsuka, C.; Ikeda, H.; Goto, Y.; Sato, Y.; Kimura, T.; Hirai, K.; Ohta, S. Downregulation of type III interferons in patients with severe COVID-19. J. Med. Virol. 2021, 93, 4559–4563. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan, P.; Andrews, J.R.; Bonilla, H.; Hedlin, H.; Jacobson, K.B.; Balasubramanian, V.; Purington, N.; Kamble, S.; de Vries, C.R.; Quintero, O. Peginterferon Lambda-1a for treatment of outpatients with uncomplicated COVID-19: A randomized placebo-controlled trial. Nat. Commun. 2021, 12, 1967. [Google Scholar] [CrossRef] [PubMed]
- Feld, J.J.; Kandel, C.; Biondi, M.J.; Kozak, R.A.; Zahoor, M.A.; Lemieux, C.; Borgia, S.M.; Boggild, A.K.; Powis, J.; McCready, J. Peginterferon lambda for the treatment of outpatients with COVID-19: A phase 2, placebo-controlled randomised trial. Lancet Respir. Med. 2021, 9, 498–510. [Google Scholar] [CrossRef]
- Generali, D.; Bosio, G.; Malberti, F.; Cuzzoli, A.; Testa, S.; Romanini, L.; Fioravanti, A.; Morandini, A.; Pianta, L.; Giannotti, G. Canakinumab as treatment for COVID-19-related pneumonia: A prospective case-control study. Int. J. Infect. Dis. 2021, 104, 433–440. [Google Scholar] [CrossRef]
- Caricchio, R.; Abbate, A.; Gordeev, I.; Meng, J.; Hsue, P.Y.; Neogi, T.; Arduino, R.; Fomina, D.; Bogdanov, R.; Stepanenko, T. Effect of canakinumab vs placebo on survival without invasive mechanical ventilation in patients hospitalized with severe COVID-19: A randomized clinical trial. JAMA 2021, 326, 230–239. [Google Scholar] [CrossRef]
- Tharaux, P.-L.; Pialoux, G.; Pavot, A.; Mariette, X.; Hermine, O.; Resche-Rigon, M.; Porcher, R.; Ravaud, P.; Bureau, S.; Dougados, M. Effect of anakinra versus usual care in adults in hospital with COVID-19 and mild-to-moderate pneumonia (CORIMUNO-ANA-1): A randomised controlled trial. Lancet Respir. Med. 2021, 9, 295–304. [Google Scholar] [CrossRef]
- Kyriazopoulou, E.; Panagopoulos, P.; Metallidis, S.; Dalekos, G.N.; Poulakou, G.; Gatselis, N.; Karakike, E.; Saridaki, M.; Loli, G.; Stefos, A. An open label trial of anakinra to prevent respiratory failure in COVID-19. eLife 2021, 10, e66125. [Google Scholar] [CrossRef]
- Pontali, E.; Volpi, S.; Signori, A.; Antonucci, G.; Castellaneta, M.; Buzzi, D.; Montale, A.; Bustaffa, M.; Angelelli, A.; Caorsi, R. Efficacy of early anti-inflammatory treatment with high doses of intravenous anakinra with or without glucocorticoids in patients with severe COVID-19 pneumonia. J. Allergy Clin. Immunol. 2021, 147, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Mattoo, S.-u.-S.; Myoung, J. T cell responses to SARS-CoV-2 in humans and animals. J. Microbiol. 2022, 60, 1–14. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| SARS-CoV-2 Protein | Inhibitory Effect on Immune System | Mechanism of Action | Reference |
|---|---|---|---|
| NSP1 | mRNA export | Prevention of adequate NXF1 and host mRNA interaction | [55] |
| Protein translation | Interaction with the 40S ribosomal subunit | ||
| Inhibits Type I IFN signaling pathway | Inhibition of the phosphorylation of STAT1 and STAT2 | ||
| NSP3 | Recognition by PRRs | DMV formation, helping to escape recognition of vRNA by molecular sensors | [46,47,48,63] |
| Protein degradation | Cleavage of IRF3 through papain-like protease activity | ||
| Type I IFN synthesis | Prevention of IRF3 phosphorylation, dimerization, and translocation | ||
| NSP4 | Recognition by PRRs | DMV formation | [46,47,48] |
| NSP6 | Recognition by PRRs | DMV formation | [46,47,48,68] |
| Type I IFN synthesis | Inhibition of IRF3 phosphorylation | ||
| Type I IFN signaling pathway | Inhibition of STAT1 and STAT2 phosphorylation | ||
| NSP8 | Type I IFN synthesis | Impairment of K63-linked polyubiquitination of MDA5 | [61,71] |
| Protein trafficking | Binding to 7SLRNA | ||
| NSP9 | Protein trafficking | Binding to 7SLRNA | |
| NSP12 | Type I IFN signaling pathway | Inhibition of the nuclear translocation of IRF3 | [72] |
| NSP13 | Recognition by PRRs | Hydrolysis of the first phosphate from the nascent RNA | [49,68,73,74] |
| Type I IFN synthesis | Down-regulation of TBK1 recruitment to MAVS | ||
| Type I IFN signaling pathway | Inhibition of STAT1 and STAT2 phosphorylation | ||
| NSP14 | Recognition by PRRs | Methylation of capped viral RNA together with NSP10 | [46,50] |
| Protein degradation | Lysosomal degradation of IFNAR1 | ||
| NSP16 | Recognition by PRRs | Methylation of the ribose 2′-O of the first nucleotide of viral Cap-O-RNA together with NSP10 | [51,52,61] |
| Decreased ISG activity | Disruption of host mRNA splicing | ||
| N | Type I IFN synthesis | Inhibition of TRIM25-mediated RIG-I ubiquitination and activationand inhibition of TBK1-IRF3 interaction, phosphorylation, and nuclear translocation of IRF3 | [76,77,78,80] |
| Type I IFN signaling pathway | Inhibition of the phosphorylation and nuclear translocation of STAT1 and STAT2 | ||
| M | Protein degradation | Proteasomal degradation of TBK1 through K48-linked ubiquitination | [62,68,83] |
| Type I IFN synthesis | Inhibition of RIG-I—MAVS, MAVS—TBK1, TRAF3—TBK1 interactions. Downregulation of phosphorylation and nuclear translocation of IRF3 | ||
| ORF3a | Type I IFN synthesis | Inhibition of IRF3 nuclear translocation | [68,74] |
| Type I IFN signaling pathway | Inhibition of STAT1 phosphorylation | ||
| ORF3b | Type I IFN synthesis | Inhibition of IRF3 nuclear translocation by sequestering IRF3 outside the nucleus | [86] |
| ORF6 | Type I IFN synthesis | Inhibition of IRF3 nuclear translocation | [68,88,89,90] |
| Type I IFN signaling pathway | Inhibition of STAT1 nuclear translocation | ||
| ORF7a | Type I IFN signaling pathway | Inhibition of STAT1 phosphorylation | [68] |
| ORF7b | Type I IFN signaling pathway | Inhibition of STAT1 and STAT2 phosphorylation | [68] |
| ORF9b | Type I IFN synthesis | Interruption of K63-linked ubiquitination of IKKγInteraction with TOM70, presumably disrupting MAVS and TBK1/IRF3 interaction | [25,94] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mattoo, S.-u.-S.; Kim, S.-J.; Ahn, D.-G.; Myoung, J. Escape and Over-Activation of Innate Immune Responses by SARS-CoV-2: Two Faces of a Coin. Viruses 2022, 14, 530. https://doi.org/10.3390/v14030530
Mattoo S-u-S, Kim S-J, Ahn D-G, Myoung J. Escape and Over-Activation of Innate Immune Responses by SARS-CoV-2: Two Faces of a Coin. Viruses. 2022; 14(3):530. https://doi.org/10.3390/v14030530
Chicago/Turabian StyleMattoo, Sameer-ul-Salam, Seong-Jun Kim, Dae-Gyun Ahn, and Jinjong Myoung. 2022. "Escape and Over-Activation of Innate Immune Responses by SARS-CoV-2: Two Faces of a Coin" Viruses 14, no. 3: 530. https://doi.org/10.3390/v14030530
APA StyleMattoo, S.-u.-S., Kim, S.-J., Ahn, D.-G., & Myoung, J. (2022). Escape and Over-Activation of Innate Immune Responses by SARS-CoV-2: Two Faces of a Coin. Viruses, 14(3), 530. https://doi.org/10.3390/v14030530