
Early Genomic, Epidemiological, and Clinical Description of the SARS-CoV-2 Omicron Variant in Mexico City

 ,
,  , , , and add
Show full author list
, , , and add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
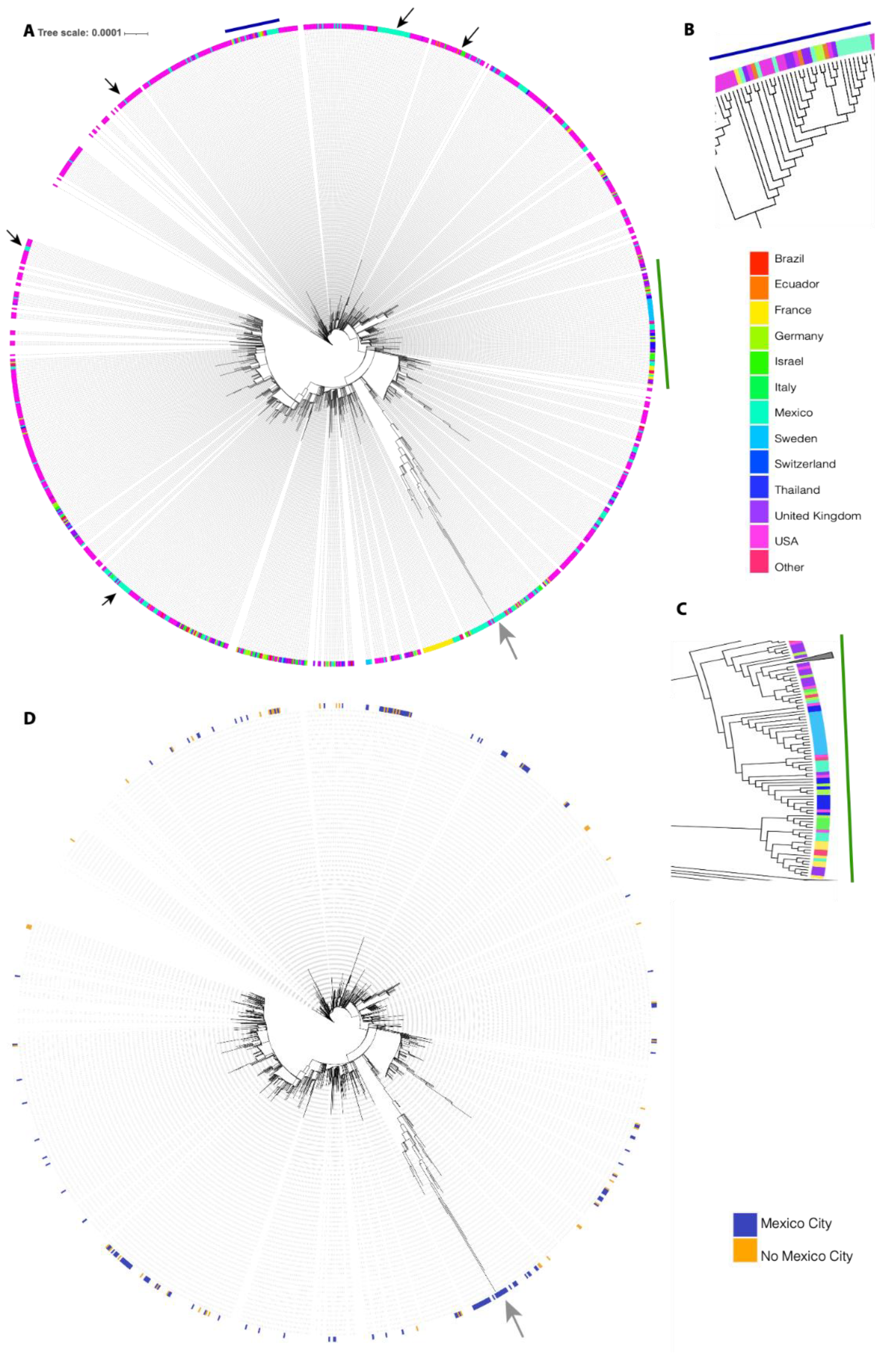
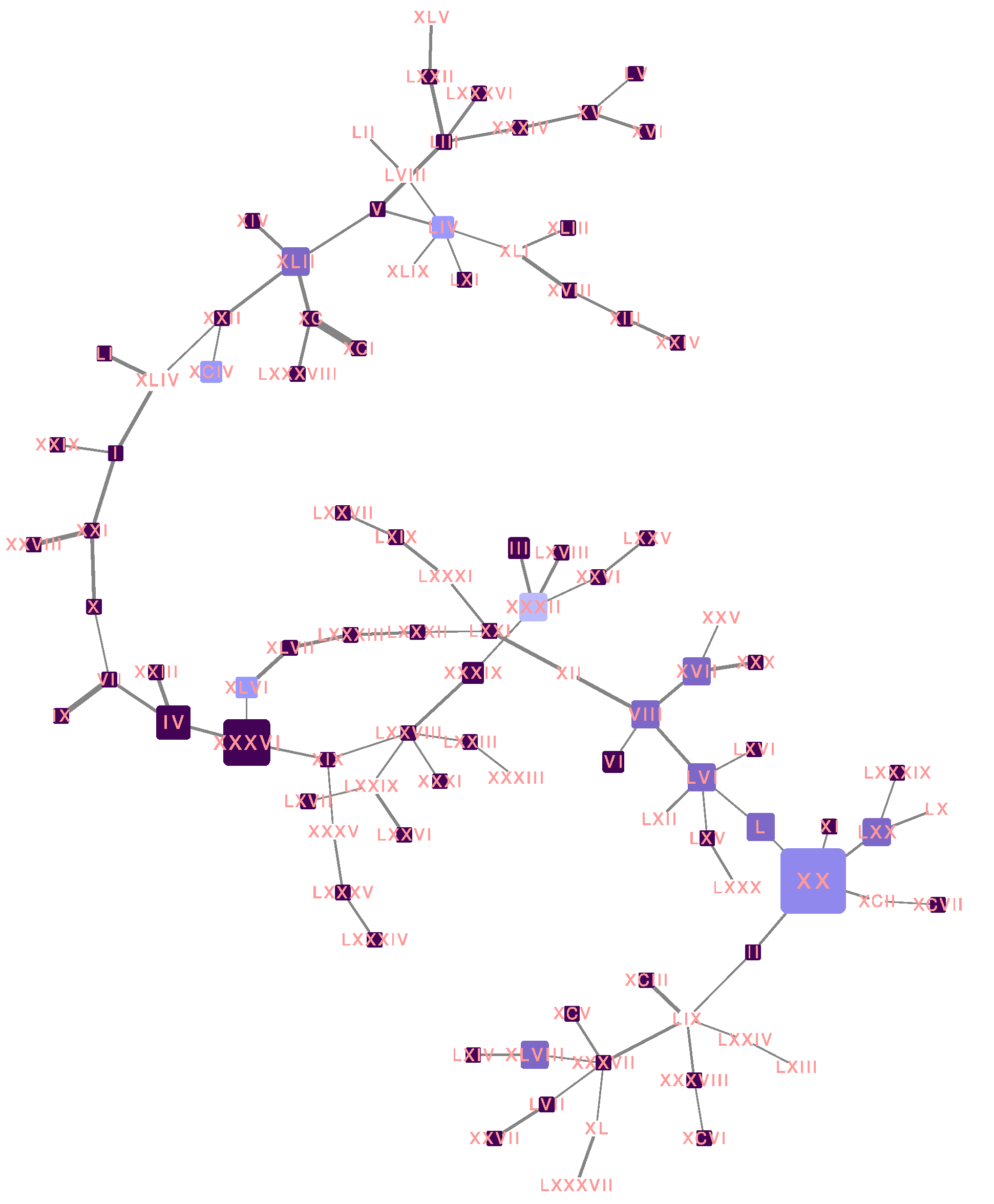
3.1. Phylogenetic Analysis
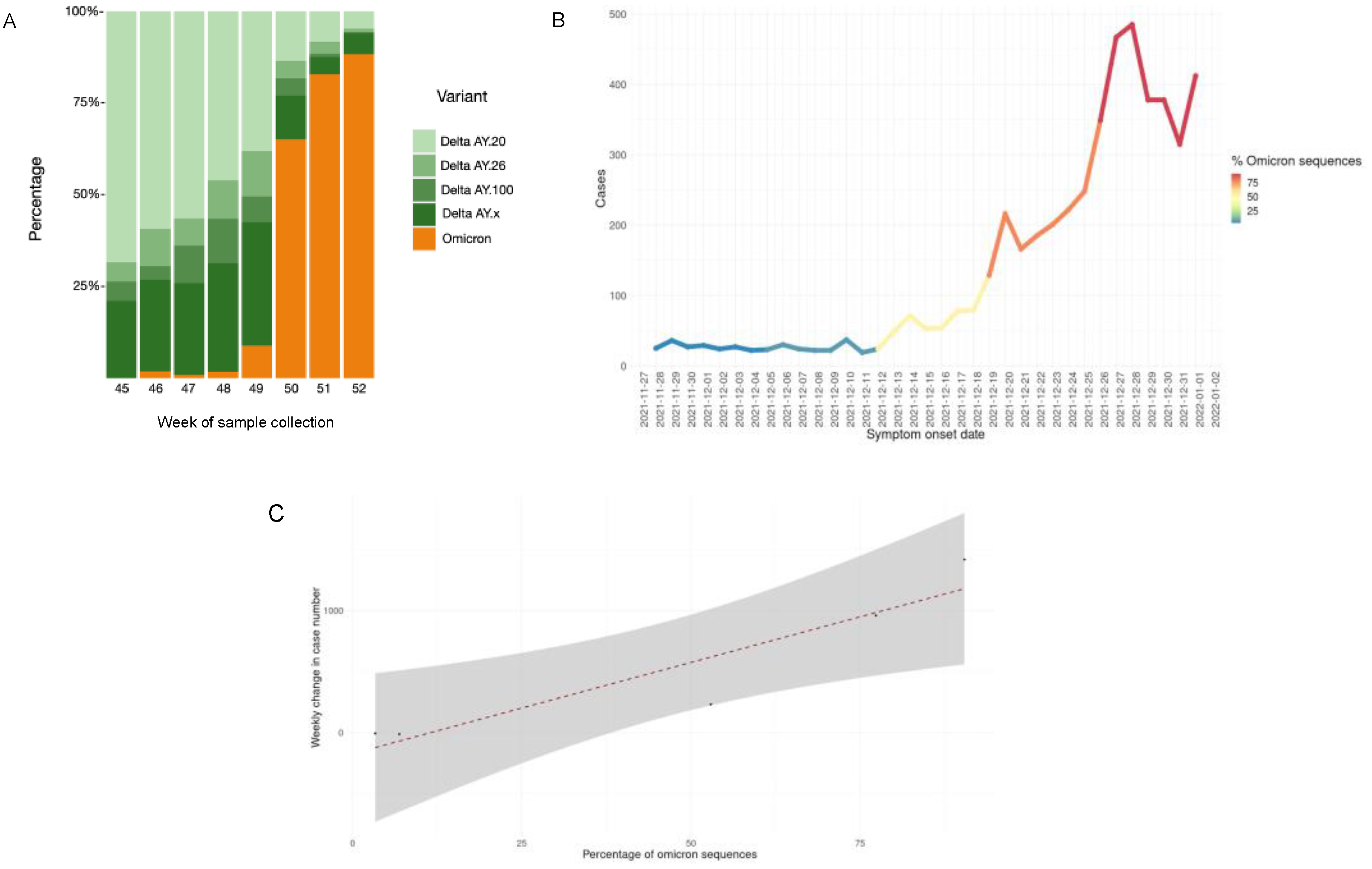
3.2. Identification of the Omicron Variant
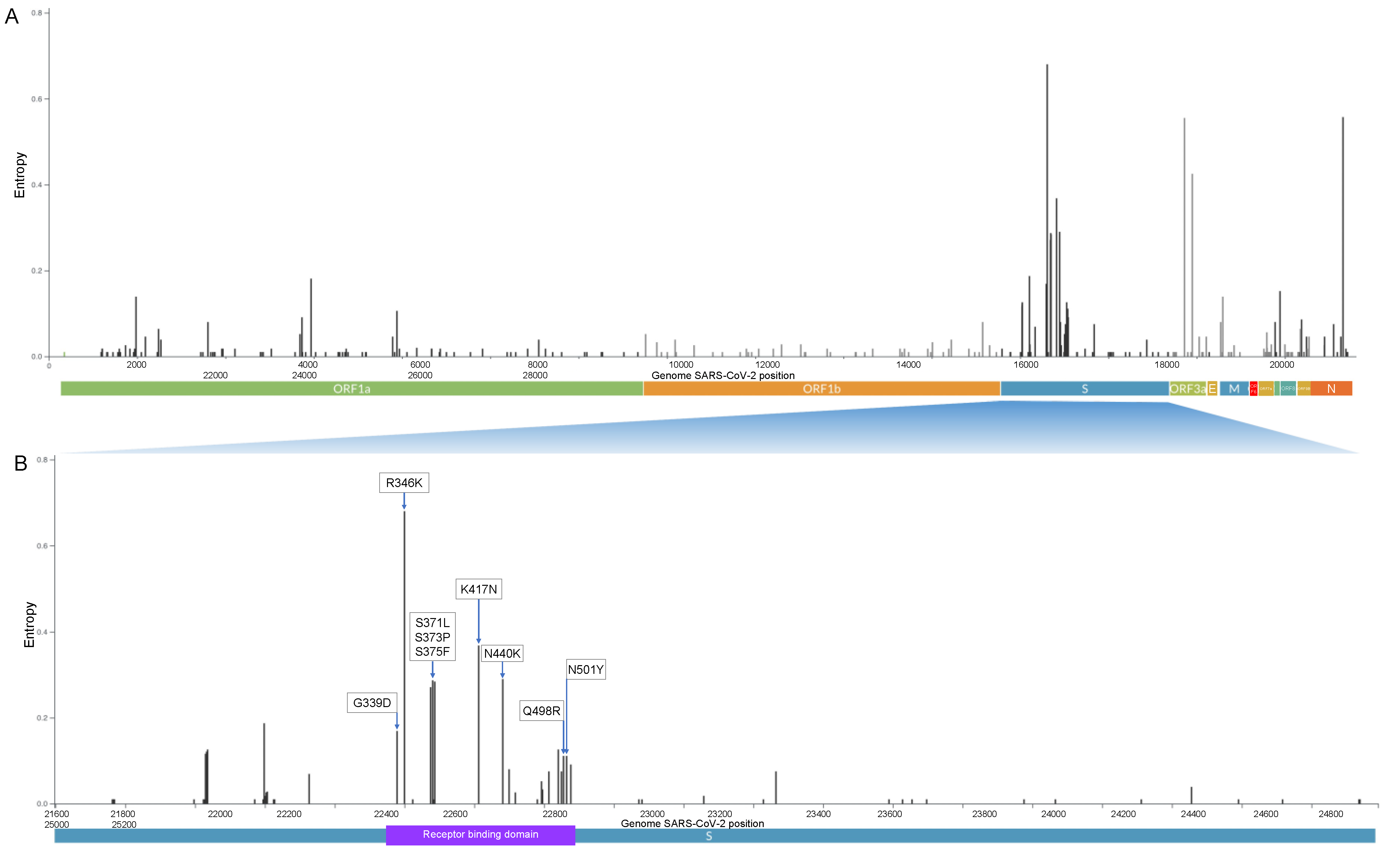
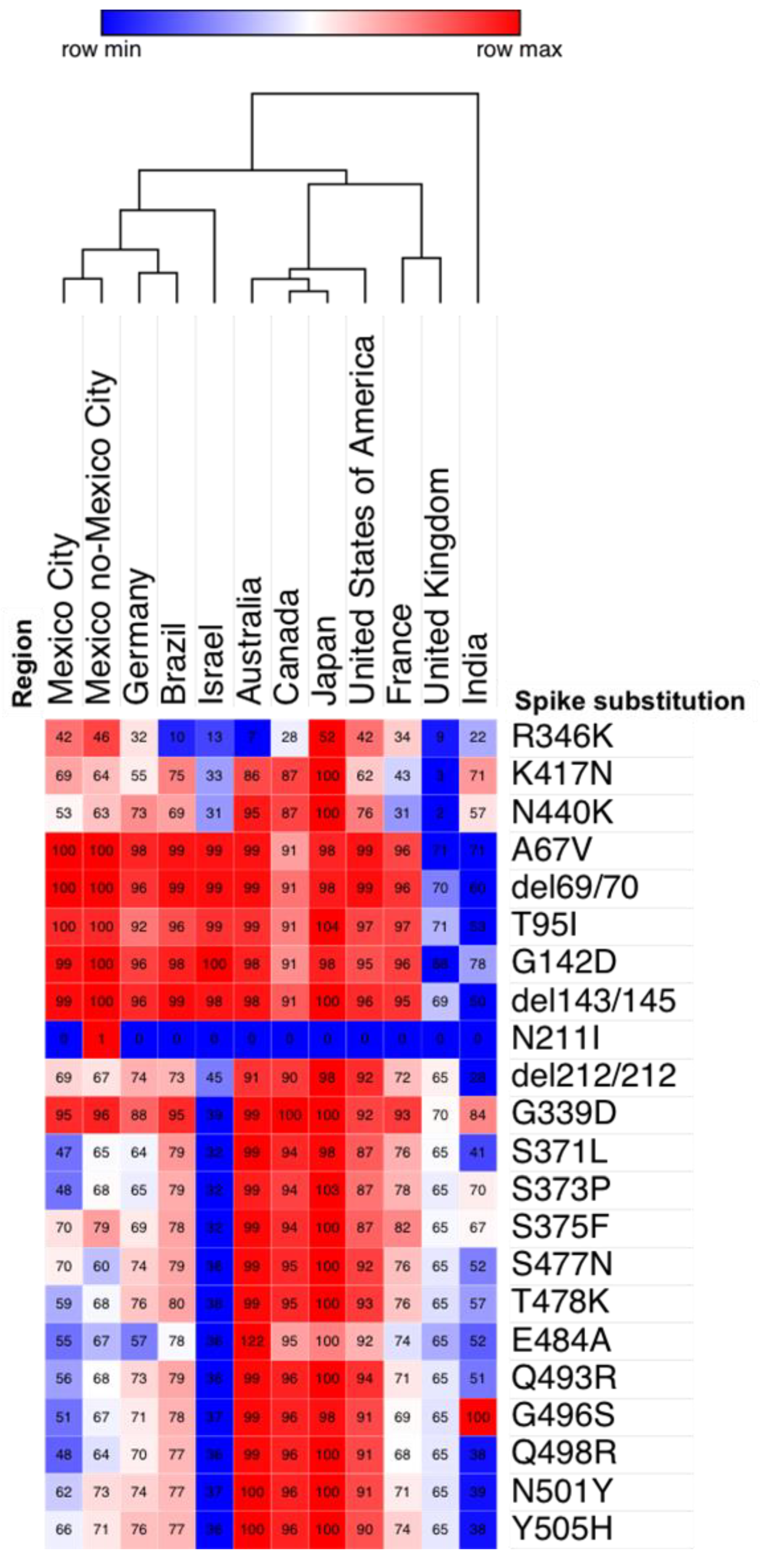
3.3. Omicron Genetic Background
3.4. Prevalence of the R346K Substitution
3.5. Haplotype Network
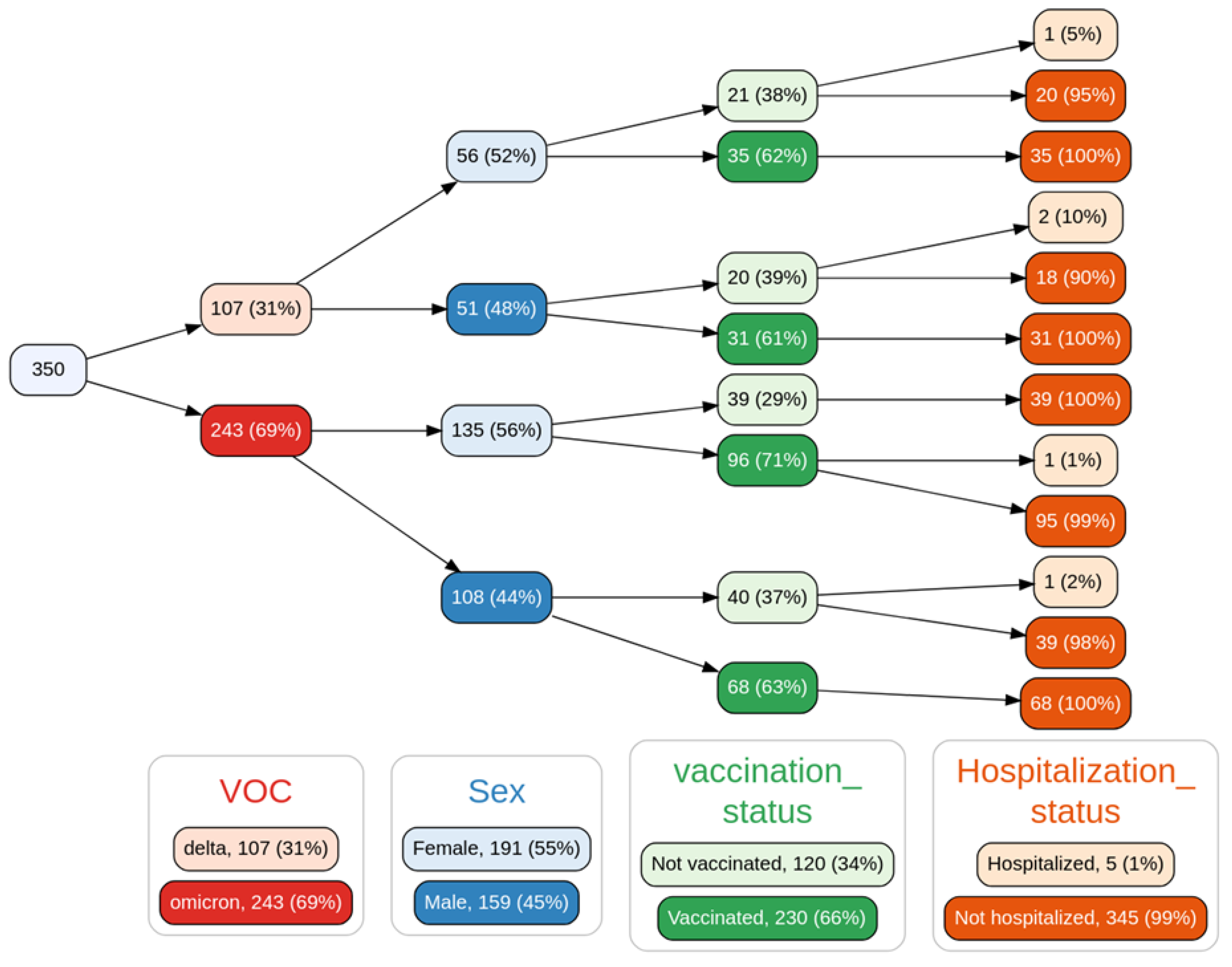
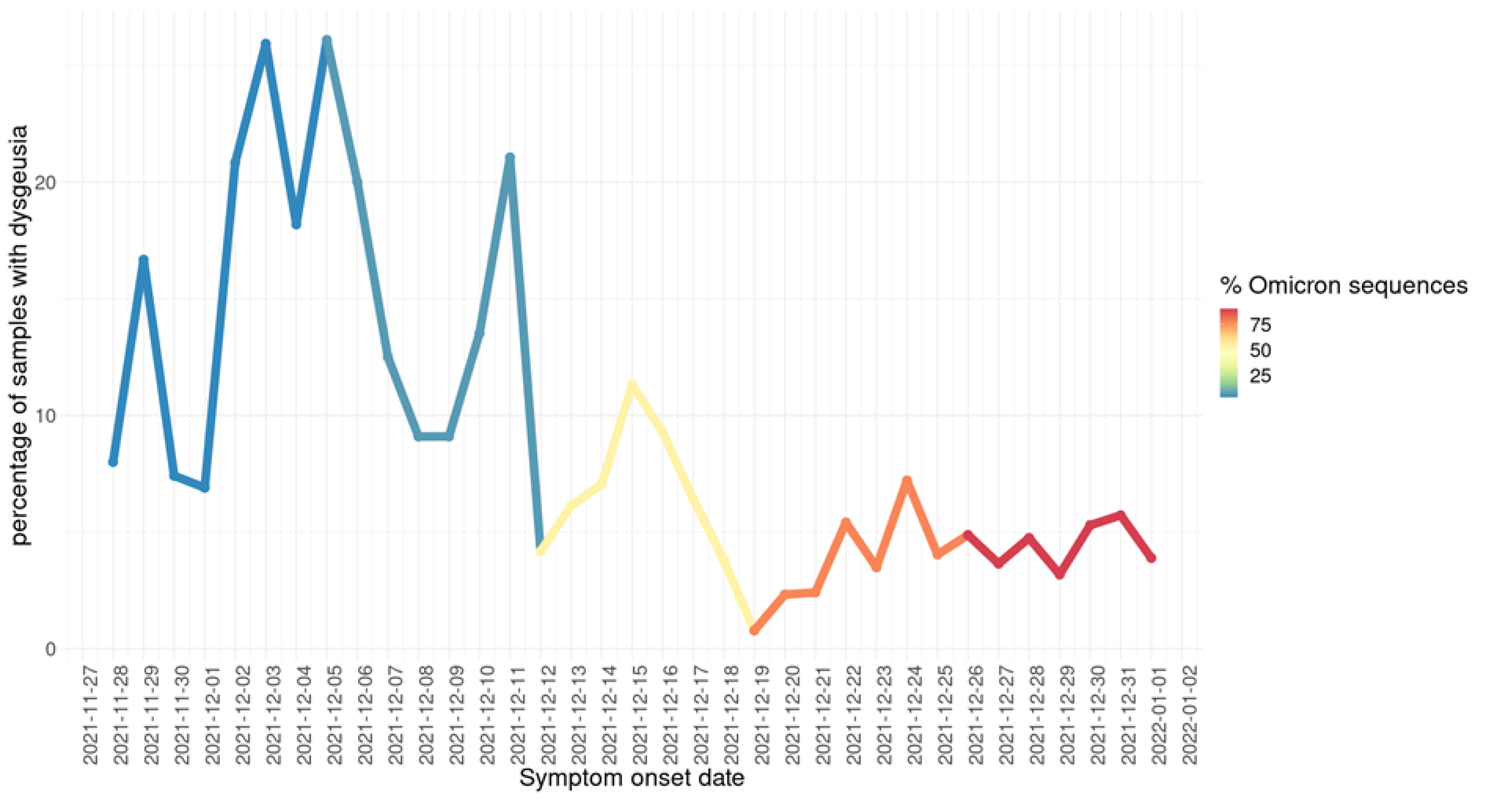
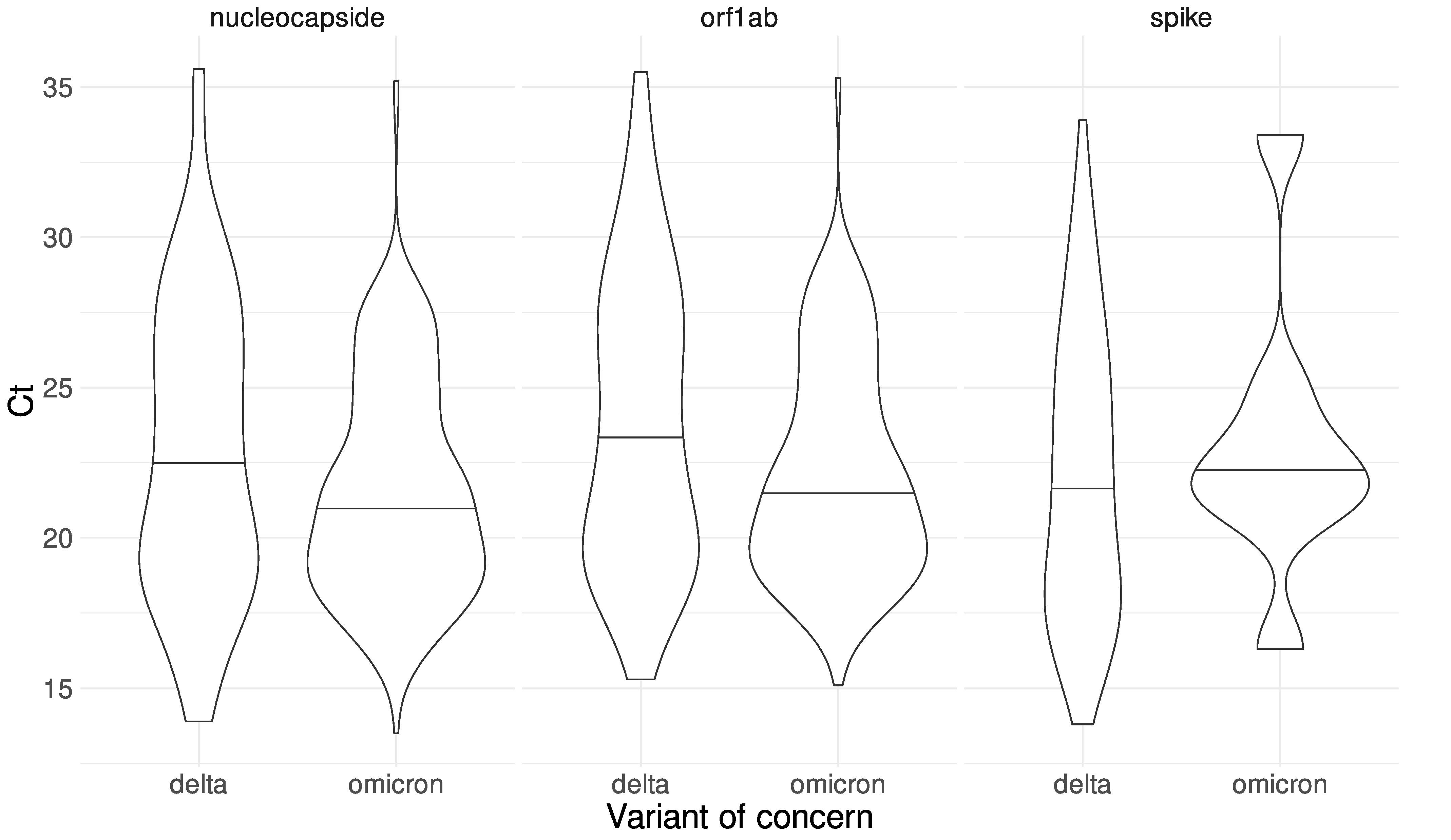
3.6. Epidemiology and Clinical Associations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tian, D.; Sun, Y.; Zhou, J.; Ye, Q. The Global Epidemic of SARS-CoV-2 Variants and Their Mutational Immune Escape. J. Med. Virol. 2022, 94, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Salehi-Vaziri, M.; Fazlalipour, M.; Seyed Khorrami, S.M.; Azadmanesh, K.; Pouriayevali, M.H.; Jalali, T.; Shoja, Z.; Maleki, A. The Ins and Outs of SARS-CoV-2 Variants of Concern (VOCs). Arch. Virol. 2022, 167, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Elbe, S.; Buckland-Merrett, G. Data, Disease and Diplomacy: GISAID’s Innovative Contribution to Global Health: Data, Disease and Diplomacy. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khandia, R.; Singhal, S.; Alqahtani, T.; Kamal, M.A.; El-Shall, N.A.; Nainu, F.; Desingu, P.A.; Dhama, K. Emergence of SARS-CoV-2 Omicron (B.1.1.529) Variant, Salient Features, High Global Health Concerns and Strategies to Counter It amid Ongoing COVID-19 Pandemic. Environ. Res. 2022, 209, 112816. [Google Scholar] [CrossRef] [PubMed]
- Sifuentes-Osornio, J.; Angulo-Guerrero, O.; De-Anda-Jáuregui, G.; Díaz-De-León-Santiago, J.L.; Hernández-Lemus, E.; Benítez-Pérez, H.; Herrera, L.A.; López-Arellano, O.; Revuelta-Herrera, A.; Rosales-Tapia, A.R.; et al. Probability of Hospitalization and Death among COVID-19 Patients with Comorbidity during Outbreaks Occurring in Mexico City. Medrxiv 2021. [Google Scholar] [CrossRef]
- Cedro-Tanda, A.; Gómez-Romero, L.; Alcaraz, N.; de Anda-Jauregui, G.; Peñaloza, F.; Moreno, B.; Escobar-Arrazola, M.A.; Ramirez-Vega, O.A.; Munguia-Garza, P.; Garcia-Cardenas, F.; et al. The Evolutionary Landscape of SARS-CoV-2 Variant B.1.1.519 and Its Clinical Impact in Mexico City. Viruses 2021, 13, 2182. [Google Scholar] [CrossRef]
- DataMEXICO. Secretaría de Economía México. Available online: https://datamexico.org/ (accessed on 31 January 2022).
- Yamamoto-Elizalde, A.Y.; Hernández-Lemus, E.; De Anda-Jáuregui, G. Diffusion Processes in Multilayer Transportation Networks: The Flight of the Coronavirus. Rev. Mex. Fís. 2020, 66, 516–524. [Google Scholar] [CrossRef]
- González-Candelas, F.; Shaw, M.-A.; Phan, T.; Kulkarni-Kale, U.; Paraskevis, D.; Luciani, F.; Kimura, H.; Sironi, M. One Year into the Pandemic: Short-Term Evolution of SARS-CoV-2 and Emergence of New Lineages. Infect. Genet. Evol. 2021, 92, 104869. [Google Scholar] [CrossRef]
- Ilumina, Inc. Illumina COVIDSeq Test. Available online: https://www.illumina.com/products/by-type/ivd-products/covidseq.html (accessed on 31 January 2022).
- Umair, M.; Ikram, A.; Salman, M.; Alam, M.M.; Badar, N.; Rehman, Z.; Tamim, S.; Khurshid, A.; Ahad, A.; Ahmad, H.; et al. Importation of SARS-CoV-2 Variant B.1.1.7 in Pakistan. J. Med. Virol. 2021, 93, 2623–2625. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Wang, W.-H.; Urbina, A.N.; Tseng, S.-P.; Lu, P.-L.; Chen, Y.-H.; Yu, M.-L.; Wang, S.-F. Importation of SARS-CoV-2 Infection Leads to Major COVID-19 Epidemic in Taiwan. Int. J. Infect. Dis. 2020, 97, 240–244. [Google Scholar] [CrossRef]
- Lemieux, J.E.; Siddle, K.J.; Shaw, B.M.; Loreth, C.; Schaffner, S.F.; Gladden-Young, A.; Adams, G.; Fink, T.; Tomkins-Tinch, C.H.; Krasilnikova, L.A.; et al. Phylogenetic Analysis of SARS-CoV-2 in the Boston Area Highlights the Role of Recurrent Importation and Superspreading Events. Epidemiology 2020. [Google Scholar] [CrossRef]
- Munnink, B.B.O.; Worp, N.; Nieuwenhuijse, D.F.; Sikkema, R.S.; Haagmans, B.; Fouchier, R.A.M.; Koopmans, M. Author Correction: The next Phase of SARS-CoV-2 Surveillance: Real-Time Molecular Epidemiology. Nat. Med. 2021, 27, 2048. [Google Scholar] [CrossRef] [PubMed]
- Vogel, M.; Augusto, G.; Chang, X.; Liu, X.; Speiser, D.; Mohsen, M.O.; Bachmann, M.F. Molecular Definition of Severe Acute Respiratory Syndrome Coronavirus 2 Receptor-binding Domain Mutations: Receptor Affinity versus Neutralization of Receptor Interaction. Allergy 2022, 77, 143–149. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Dingens, A.S.; Bloom, J.D. Complete Map of SARS-CoV-2 RBD Mutations That Escape the Monoclonal Antibody LY-CoV555 and Its Cocktail with LY-CoV016. Cell Rep. Med. 2021, 2, 100255. [Google Scholar] [CrossRef] [PubMed]
- Wibmer, C.K.; Ayres, F.; Hermanus, T.; Madzivhandila, M.; Kgagudi, P.; Oosthuysen, B.; Lambson, B.E.; de Oliveira, T.; Vermeulen, M.; van der Berg, K.; et al. SARS-CoV-2 501Y.V2 Escapes Neutralization by South African COVID-19 Donor Plasma. Nat. Med. 2021, 27, 622–625. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Arora, P.; Groß, R.; Seidel, A.; Hörnich, B.F.; Hahn, A.S.; Krüger, N.; Graichen, L.; Hofmann-Winkler, H.; Kempf, A.; et al. SARS-CoV-2 Variants B.1.351 and P.1 Escape from Neutralizing Antibodies. Cell 2021, 184, 2384–2393.e12. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, S.; Wu, B.; Yang, Q.; Chen, A.; Li, Y.; Zhang, Y.; Pan, T.; Zhang, H.; He, X. SARS-CoV-2 Omicron Strain Exhibits Potent Capabilities for Immune Evasion and Viral Entrance. Signal Transduct. Target. Ther. 2021, 6, 430. [Google Scholar] [CrossRef]
- Planas, D.; Saunders, N.; Maes, P.; Guivel-Benhassine, F.; Planchais, C.; Buchrieser, J.; Bolland, W.-H.; Porrot, F.; Staropoli, I.; Lemoine, F.; et al. Considerable Escape of SARS-CoV-2 Omicron to Antibody Neutralization. Nature 2021, 602, 671–675. [Google Scholar] [CrossRef]
- Laiton-Donato, K.; Franco-Muñoz, C.; Álvarez-Díaz, D.A.; Ruiz-Moreno, H.A.; Usme-Ciro, J.A.; Prada, D.A.; Reales-González, J.; Corchuelo, S.; Herrera-Sepúlveda, M.T.; Naizaque, J.; et al. Characterization of the Emerging B.1.621 Variant of Interest of SARS-CoV-2. Infectious Diseases (except HIV/AIDS). Infect. Genet. Evol. 2021, 95, 105038. [Google Scholar] [CrossRef]
- Liu, L.; Iketani, S.; Guo, Y.; Chan, J.F.-W.; Wang, M.; Liu, L.; Luo, Y.; Chu, H.; Huang, Y.; Nair, M.S.; et al. Striking Antibody Evasion Manifested by the Omicron Variant of SARS-CoV-2. Nature 2021, 602, 676–681. [Google Scholar] [CrossRef]
- Duty, J.A.; Kraus, T.; Zhou, H.; Zhang, Y.; Shaabani, N.; Yildiz, S.; Du, N.; Singh, A.; Miorin, L.; Li, D.; et al. Discovery of a SARS-CoV-2 Broadly-Acting Neutralizing Antibody with Activity against Omicron and Omicron + R346K Variants. bioRxiv 2022. [Google Scholar] [CrossRef]
- Cameroni, E.; Bowen, J.E.; Rosen, L.E.; Saliba, C.; Zepeda, S.K.; Culap, K.; Pinto, D.; VanBlargan, L.A.; De Marco, A.; di Iulio, J.; et al. Broadly Neutralizing Antibodies Overcome SARS-CoV-2 Omicron Antigenic Shift. Nature 2021, 602, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, N.; Hamaguchi, M. The Importance of the Genetic Diversity of the HcRNAV SsRNA Virus in the Viral-Based Bloom Control of the Dinoflagellate Heterocapsa Circularisquama. Aquaculture 2022, 546, 737318. [Google Scholar] [CrossRef]
- Chaw, S.-M.; Tai, J.-H.; Chen, S.-L.; Hsieh, C.-H.; Chang, S.-Y.; Yeh, S.-H.; Yang, W.-S.; Chen, P.-J.; Wang, H.-Y. The Origin and Underlying Driving Forces of the SARS-CoV-2 Outbreak. J. Biomed. Sci. 2020, 27, 73. [Google Scholar] [CrossRef] [PubMed]
- Bui, N.-N.; Lin, Y.-T.; Huang, S.-H.; Lin, C.-W. Haplotype Distribution of SARS-CoV-2 Variants in Low and High Vaccination Rate Countries during Ongoing Global COVID-19 Pandemic in Early 2021. Infect. Genet. Evol. 2022, 97, 105164. [Google Scholar] [CrossRef] [PubMed]
- Veneti, L.; Bøås, H.; Kristoffersen, A.B.; Stålcrantz, J.; Bragstad, K.; Hungnes, O.; Storm, M.L.; Aasand, N.; Rø, G.; Starrfelt, J.; et al. Reduced Risk of Hospitalisation among Reported COVID-19 Cases Infected with the SARS-CoV-2 Omicron BA.1 Variant Compared with the Delta Variant, Norway, December 2021 to January 2022. Eurosurveillance 2022, 27, 2200077. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, R.P.; Hanage, W.P. Challenges in Inferring Intrinsic Severity of the SARS-CoV-2 Omicron Variant. N. Engl. J. Med. 2022, 386, e14. [Google Scholar] [CrossRef]
- Bello-Chavolla, O.Y.; Bahena-López, J.P.; Antonio-Villa, N.E.; Vargas-Vázquez, A.; González-Díaz, A.; Márquez-Salinas, A.; Fermín-Martínez, C.A.; Naveja, J.J.; Aguilar-Salinas, C.A. Predicting Mortality due to SARS-CoV-2: A Mechanistic Score Relating Obesity and Diabetes to COVID-19 Outcomes in Mexico. J. Clin. Endocrinol. Metab. 2020, 105, 2752–2761. [Google Scholar] [CrossRef]
- UK Health Security Agency. SARS-CoV-2 Variants of Concern and Variants under Investigation in England. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1057359/Technical-Briefing-37-25February2022.pdf (accessed on 1 February 2022).
- Dahdouh, E.; Lázaro-Perona, F.; Romero-Gómez, M.P.; Mingorance, J.; García-Rodriguez, J. Ct Values from SARS-CoV-2 Diagnostic PCR Assays Should Not Be Used as Direct Estimates of Viral Load. J. Infect. 2021, 82, 414–451. [Google Scholar] [CrossRef]
- Michalakis, Y.; Sofonea, M.T.; Alizon, S.; Bravo, I.G. SARS-CoV-2 Viral RNA Levels Are Not “Viral Load”. Trends Microbiol. 2021, 29, 970–972. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | p-Value |
|---|---|
| Sex | 0.6414 |
| Vax | 0.3285 |
| Hospitalization status | 0.1692 |
| Age | 0.1801 |
| Number of comorbidities | 0.7300 |
| Number of symptoms | 0.8343 |
| Term | Odds Ratio | p-Value |
|---|---|---|
| Intercept | 2.01 | 0.0021 |
| Odynophagia | 2.10 | 0.0090 |
| Dysgeusia | 0.103 | 0.0009 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cedro-Tanda, A.; Gómez-Romero, L.; de Anda-Jauregui, G.; Garnica-López, D.; Alfaro-Mora, Y.; Sánchez-Xochipa, S.; García-García, E.F.; Mendoza-Vargas, A.; Frías-Jiménez, E.J.; Moreno, B.; et al. Early Genomic, Epidemiological, and Clinical Description of the SARS-CoV-2 Omicron Variant in Mexico City. Viruses 2022, 14, 545. https://doi.org/10.3390/v14030545
Cedro-Tanda A, Gómez-Romero L, de Anda-Jauregui G, Garnica-López D, Alfaro-Mora Y, Sánchez-Xochipa S, García-García EF, Mendoza-Vargas A, Frías-Jiménez EJ, Moreno B, et al. Early Genomic, Epidemiological, and Clinical Description of the SARS-CoV-2 Omicron Variant in Mexico City. Viruses. 2022; 14(3):545. https://doi.org/10.3390/v14030545
Chicago/Turabian StyleCedro-Tanda, Alberto, Laura Gómez-Romero, Guillermo de Anda-Jauregui, Dora Garnica-López, Yair Alfaro-Mora, Sonia Sánchez-Xochipa, Eulices F. García-García, Alfredo Mendoza-Vargas, Emmanuel J. Frías-Jiménez, Bernardo Moreno, and et al. 2022. "Early Genomic, Epidemiological, and Clinical Description of the SARS-CoV-2 Omicron Variant in Mexico City" Viruses 14, no. 3: 545. https://doi.org/10.3390/v14030545
APA StyleCedro-Tanda, A., Gómez-Romero, L., de Anda-Jauregui, G., Garnica-López, D., Alfaro-Mora, Y., Sánchez-Xochipa, S., García-García, E. F., Mendoza-Vargas, A., Frías-Jiménez, E. J., Moreno, B., Campos-Romero, A., Moreno-Camacho, J. L., Alcantar-Fernández, J., Ortíz-Ramírez, J., Benitez-González, M., Trejo-González, R., Aguirre-Chavarría, D., Núñez-Martínez, M. E., Uribe-Figueroa, L., ... Herrera, L. A. (2022). Early Genomic, Epidemiological, and Clinical Description of the SARS-CoV-2 Omicron Variant in Mexico City. Viruses, 14(3), 545. https://doi.org/10.3390/v14030545