
A BioID-Derived Proximity Interactome for SARS-CoV-2 Proteins



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmids
2.2. Cell Culture
2.3. Immunofluorescence
2.4. Western Blot Analysis
2.5. Sample Preparation
2.6. Affinity Purification of Biotinylated Proteins
2.7. Mass Spectrometry
2.8. Data Analysis
2.9. Global Proteome Data Analysis
2.10. Network Analysis of SARS-CoV-2 Interactors
2.11. Virus-Centric Analysis of SARS-CoV-2 Interactors
2.12. Integrated Analysis of Global Proteome and PPI Data for NSP7/8/12
2.13. Integrated Analysis of SARS-CoV-2 Interactome Datasets
2.14. Web-Based Shiny App
3. Results
3.1. Development of Stable BioID Cell Lines Expressing Individual SARS-CoV-2 Viral Proteins
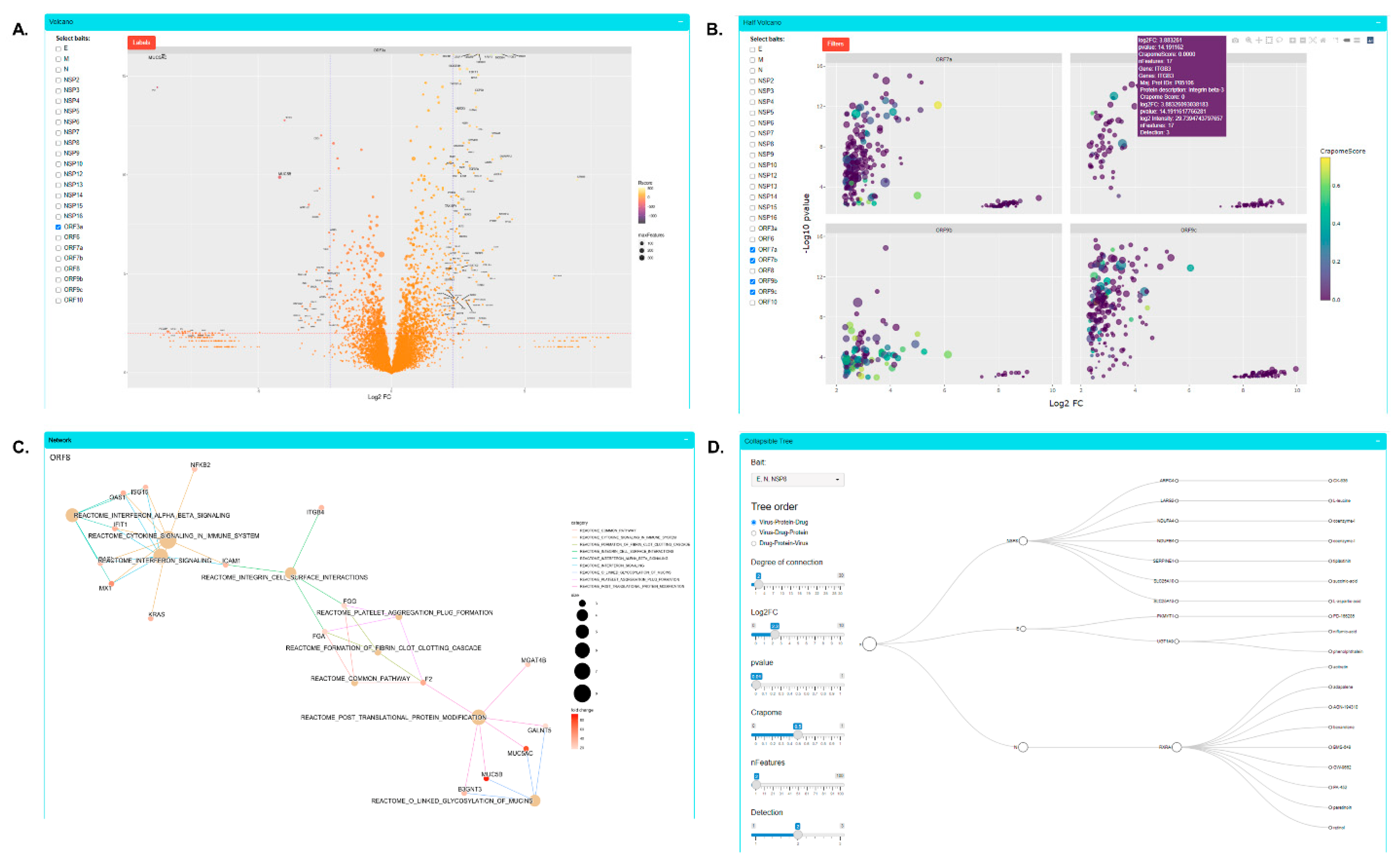
3.2. SARS-CoV-2 Proteomics Website
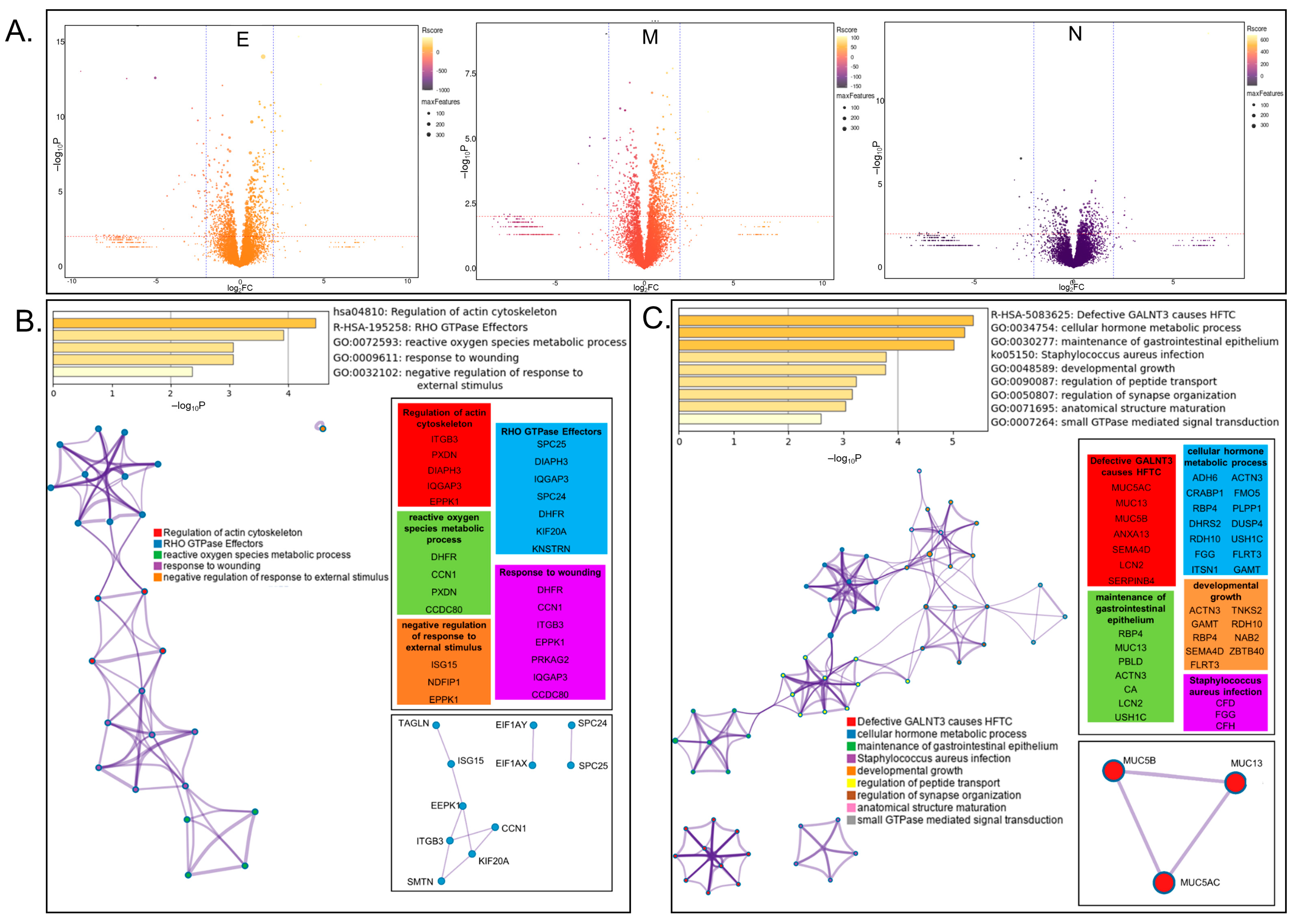
3.3. Whole-Proteome Analysis of Cells Overexpressing Individual BioID-Viral Bait Fusion Proteins
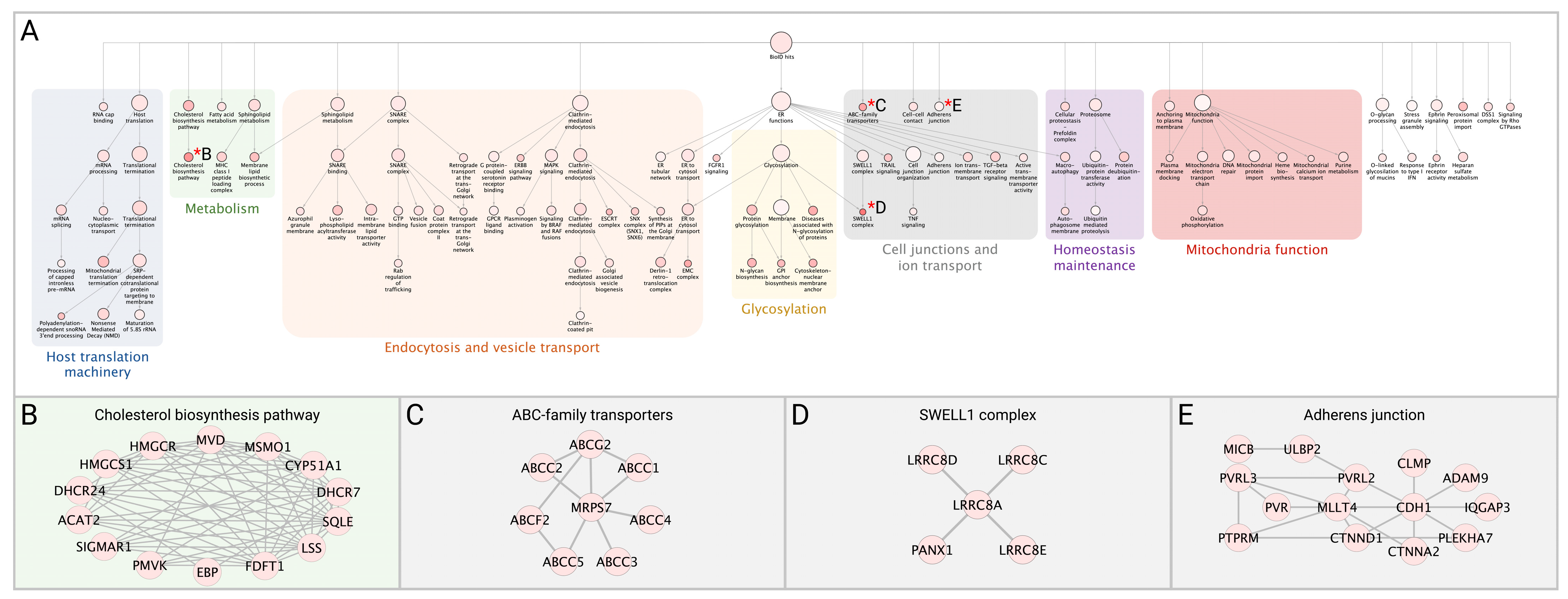
3.4. Network Analysis of SARS-CoV-2 Host Interactors Reveals Novel Biology
3.5. Focused Analysis of Individual Viral–Host Protein Interactions
3.5.1. ORF3a
3.5.2. ORF6
3.5.3. ORF8
3.5.4. NSP4
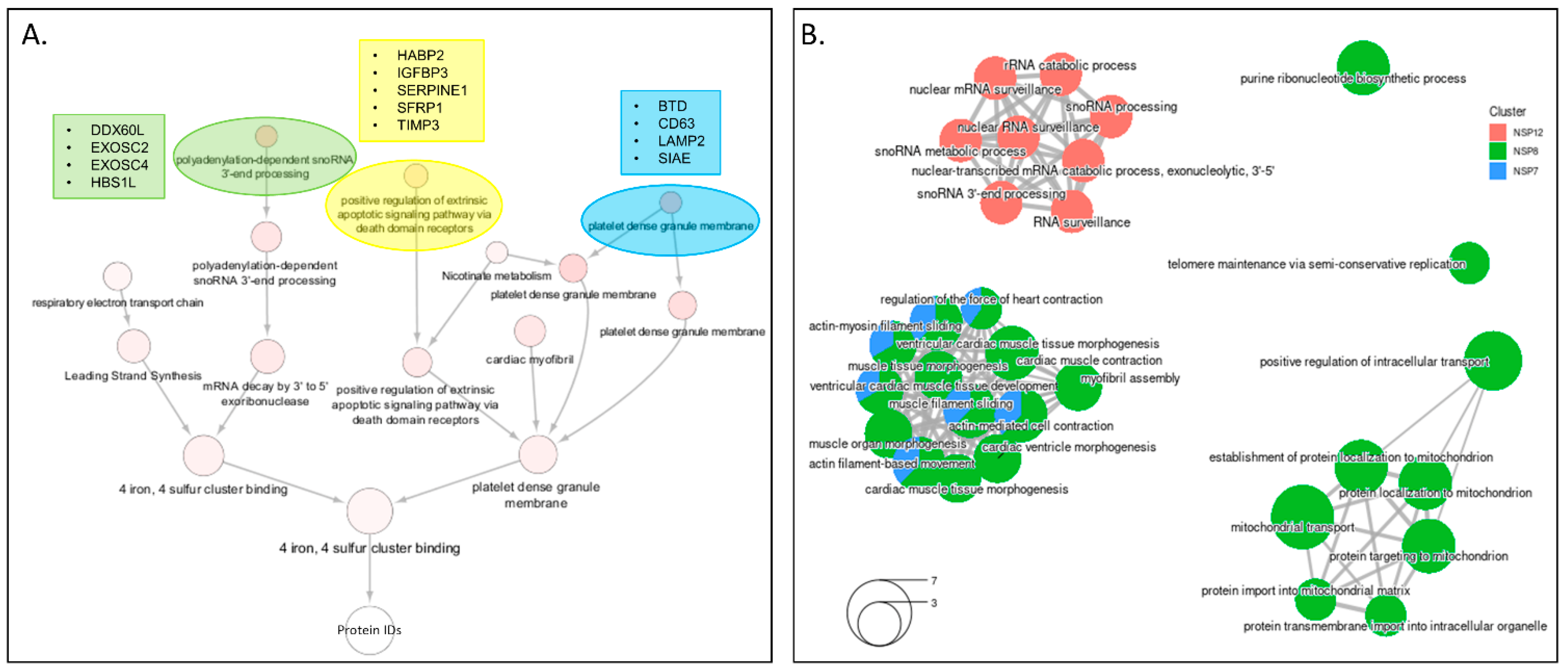
3.6. Integrated Analysis of PPI Networks and Global Abundance Changes among the Viral Proteins That Make up the RdRp Replication Complex
3.7. Integrated Analysis with Previously Published Datasets
3.7.1. SARS-CoV-2 Interaction with the Cellular Restrictome
3.7.2. Utilizing Previous SARS-CoV-2 BioID Interactome Datasets to Develop a List of High-Confidence Interactions
3.7.3. Cross-Referencing PPI Interactions with the CLUE Drug Library for Drug Repurposing Efforts
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Davies, D.A.; Adlimoghaddam, A.; Albensi, B.C. The Effect of COVID-19 on NF-kappaB and Neurological Manifestations of Disease. Mol. Neurobiol. 2021, 58, 4178–4187. [Google Scholar] [CrossRef]
- De Lorenzo, A.; Ab Kasal, D.; Tura, B.R.; Lamas, C.C.; Rey, H.C. Acute cardiac injury in patients with COVID-19. Am. J. Cardiovasc Dis. 2020, 10, 28–33. [Google Scholar] [PubMed]
- Villapol, S. Gastrointestinal symptoms associated with COVID-19: Impact on the gut microbiome. Transl. Res. 2020, 226, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Maltezou, H.; Pavli, A.; Tsakris, A. Post-COVID Syndrome: An Insight on Its Pathogenesis. Vaccines 2021, 9, 497. [Google Scholar] [CrossRef]
- Orrù, G.; Bertelloni, D.; Diolaiuti, F.; Mucci, F.; Di Giuseppe, M.; Biella, M.; Gemignani, A.; Ciacchini, R.; Conversano, C. Long-COVID Syndrome? A Study on the Persistence of Neurological, Psychological and Physiological Symptoms. Healthcare 2021, 9, 575. [Google Scholar] [CrossRef] [PubMed]
- Saadaoui, M.; Kumar, M.; Al Khodor, S. COVID-19 Infection during Pregnancy: Risk of Vertical Transmission, Fetal, and Neonatal Outcomes. J. Pers. Med. 2021, 11, 483. [Google Scholar] [CrossRef] [PubMed]
- Bari, E.; Ferrarotti, I.; Saracino, L.; Perteghella, S.; Torre, M.; Richeldi, L.; Corsico, A. Mesenchymal Stromal Cell Secretome for Post-COVID-19 Pulmonary Fibrosis: A New Therapy to Treat the Long-Term Lung Sequelae? Cells 2021, 10, 1203. [Google Scholar] [CrossRef] [PubMed]
- Tenforde, M.W.; Kim, S.S.; Lindsell, C.J.; Rose, E.B.; Shapiro, N.I.; Files, D.C.; Gibbs, K.W.; Erickson, H.L.; Steingrub, J.S.; Smithline, H.A.; et al. Symptom Duration and Risk Factors for Delayed Return to Usual Health Among Outpatients with COVID-19 in a Multistate Health Care Systems Network—United States, March–June 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 993–998. [Google Scholar] [CrossRef]
- Logue, J.K.; Franko, N.M.; McCulloch, D.J.; McDonald, D.; Magedson, A.; Wolf, C.R.; Chu, H.Y. Sequelae in Adults at 6 Months After COVID-19 Infection. JAMA Netw. Open 2021, 4, e210830. [Google Scholar] [CrossRef]
- Pavli, A.; Theodoridou, M.; Maltezou, H.C. Post-COVID Syndrome: Incidence, Clinical Spectrum, and Challenges for Primary Healthcare Professionals. Arch. Med. Res. 2021, 52, 575–581. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Mishra, B.; Mehmood, A.; Athar, M.; Mukhtar, M.S. Integrative Network Biology Framework Elucidates Molecular Mechanisms of SARS-CoV-2 Pathogenesis. iScience 2020, 23, 101526. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Guo, M.; Tian, X.; Wang, X.; Yang, X.; Wu, P.; Liu, C.; Xiao, Z.; Qu, Y.; Yin, Y.; et al. Virus-Host Interactome and Proteomic Survey Reveal Potential Virulence Factors Influencing SARS-CoV-2 Pathogenesis. Med 2020, 2, 99–112.e7. [Google Scholar] [CrossRef] [PubMed]
- Laurent, E.M.N.; Sofianatos, Y.; Komarova, A.; Gimeno, J.-P.; Tehrani, P.S.; Kim, D.-K.; Abdouni, H.; Duhamel, M.; Cassonnet, P.; Knapp, J.J.; et al. Global BioID-based SARS-CoV-2 proteins proximal interactome unveils novel ties between viral polypeptides and host factors involved in multiple COVID19-associated mechanisms. bioRxiv 2020. [Google Scholar] [CrossRef]
- Samavarchi-Tehrani, P.; Abdouni, H.; Knight, J.D.R.; Astori, A.; Samson, R.; Lin, Z.-Y.; Kim, D.-K.; Knapp, J.J.; St-Germain, J.; Go, C.D.; et al. A SARS-CoV-2–host proximity interactome. bioRxiv 2020. [Google Scholar] [CrossRef]
- Stukalov, A.; Girault, V.; Grass, V.; Karayel, O.; Bergant, V.; Urban, C.; Haas, D.A.; Huang, Y.; Oubraham, L.; Wang, A.; et al. Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV. Nature 2021, 594, 246–252. [Google Scholar] [CrossRef]
- Davies, P.P.; Almasy, P.M.; McDonald, P.F.; Plate, P. Comparative multiplexed interactomics of SARS-CoV-2 and homologous coronavirus non-structural proteins identifies unique and shared host-cell dependencies. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hoffmann, H.-H.; Sánchez-Rivera, F.J.; Schneider, W.M.; Luna, J.M.; Soto-Feliciano, Y.M.; Ashbrook, A.W.; le Pen, J.; Leal, A.A.; Ricardo-Lax, I.; Michailidis, E.; et al. Functional interrogation of a SARS-CoV-2 host protein interactome identifies unique and shared coronavirus host factors. Cell Host Microbe 2021, 29, 267–280.e5. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, C.; Feng, X.; Nie, L.; Tang, M.; Zhang, H.; Xiong, Y.; Swisher, S.K.; Srivastava, M.; Chen, J. Interactomes of SARS-CoV-2 and human coronaviruses reveal host factors potentially affecting pathogenesis. EMBO J. 2021, 40, e107776. [Google Scholar] [CrossRef]
- Meyers, J.M.; Ramanathan, M.; Shanderson, R.L.; Beck, A.; Donohue, L.; Ferguson, I.; Guo, M.G.; Rao, D.S.; Miao, W.; Reynolds, D.; et al. The proximal proteome of 17 SARS-CoV-2 proteins links to disrupted antiviral signaling and host translation. bioRxiv 2021. [Google Scholar] [CrossRef]
- Terracciano, R.; Preianò, M.; Fregola, A.; Pelaia, C.; Montalcini, T.; Savino, R. Mapping the SARS-CoV-2-Host Protein-Protein Interactome by Affinity Purification Mass Spectrometry and Proximity-Dependent Biotin Labeling: A Rational and Straightforward Route to Discover Host-Directed Anti-SARS-CoV-2 Therapeutics. Int. J. Mol. Sci. 2021, 22, 532. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Huuskonen, S.; Laitinen, T.; Redchuk, T.; Bogacheva, M.; Salokas, K.; Pöhner, I.; Öhman, T.; Tonduru, A.K.; Hassinen, A.; et al. SARS-CoV-2-host proteome interactions for antiviral drug discovery. Mol. Syst. Biol. 2021, 17, e10396. [Google Scholar] [CrossRef] [PubMed]
- Boruchowicz, H.; Hawkins, J.; Cruz-Palomar, K.; Lippé, R. The XPO6 exportin mediates HSV-1 gM nuclear release late in infection. J. Virol. 2020, 94, e00753-20. [Google Scholar] [CrossRef] [PubMed]
- Coyaud, E.; Ranadheera, C.; Cheng, D.T.; Gonçalves, J.; Dyakov, B.; Laurent, E.M.; St-Germain, J.R.; Pelletier, L.; Gingras, A.-C.; Brumell, J.H.; et al. Global Interactomics Uncovers Extensive Organellar Targeting by Zika Virus. Mol. Cell. Proteom. 2018, 17, 2242–2255. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Liu, X.; Mao, F.; Yue, X.; Lee, I.; Xu, Y. Proximity proteomics identifies novel function of Rab14 in trafficking of Ebola virus matrix protein VP40. Biochem. Biophys. Res. Commun. 2020, 527, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Rider, M.A.; Cheerathodi, M.R.; Hurwitz, S.N.; Nkosi, D.; Howell, L.A.; Tremblay, D.C.; Liu, X.; Zhu, F.; Meckes, D.G. The interactome of EBV LMP1 evaluated by proximity-based BioID approach. Virology 2018, 516, 55–70. [Google Scholar] [CrossRef] [PubMed]
- V’kovski, P.; Gerber, M.; Kelly, J.; Pfaender, S.; Ebert, N.; Lagache, S.B.; Simillion, C.; Portmann, J.; Stalder, H.; Gaschen, V.; et al. Determination of host proteins composing the microenvironment of coronavirus replicase complexes by proximity-labeling. Elife 2019, 8, e42037. [Google Scholar] [CrossRef]
- May, D.G.; Scott, K.L.; Campos, A.R.; Roux, K.J. Comparative Application of BioID and TurboID for Protein-Proximity Biotinylation. Cells 2020, 9, 1070. [Google Scholar] [CrossRef]
- Pratt, D.; Chen, J.; Pillich, R.; Rynkov, V.; Gary, A.; Demchak, B.; Ideker, T. NDEx 2.0: A Clearinghouse for Research on Cancer Pathways. Cancer Res. 2017, 77, e58–e61. [Google Scholar] [CrossRef] [Green Version]
- Pratt, D.; Chen, J.; Welker, D.; Rivas, R.; Pillich, R.; Rynkov, V.; Ono, K.; Miello, C.; Hicks, L.; Szalma, S.; et al. NDEx, the Network Data Exchange. Cell Syst. 2015, 1, 302–305. [Google Scholar] [CrossRef] [Green Version]
- Mellacheruvu, D.; Wright, Z.; Couzens, A.L.; Lambert, J.-P.; St-Denis, N.A.; Li, T.; Miteva, Y.V.; Hauri, S.; Sardiu, M.E.; Low, T.Y.; et al. The CRAPome: A contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 2013, 10, 730–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlin, D.E.; Demchak, B.; Pratt, D.; Sage, E.; Ideker, T. Network propagation in the cytoscape cyberinfrastructure. PLoS Comput. Biol. 2017, 13, e1005598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, F.; Zhang, S.; Churas, C.; Pratt, D.; Bahar, I.; Ideker, T. HiDeF: Identifying persistent structures in multiscale ‘omics data. Genome Biol. 2021, 22, 21. [Google Scholar] [CrossRef] [PubMed]
- Singhal, A.; Cao, S.; Churas, C.; Pratt, D.; Fortunato, S.; Zheng, F.; Ideker, T. Multiscale community detection in Cytoscape. PLoS Comput Biol. 2020, 16, e1008239. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.K.; Kramer, M.; Dutkowski, J.; Srivas, R.; Licon, K.; Kreisberg, J.F.; Ng, C.T.; Krogan, N.; Sharan, R.; Ideker, T. Translation of Genotype to Phenotype by a Hierarchy of Cell Subsystems. Cell Syst. 2016, 2, 77–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, M.; Dutkowski, J.; Yu, M.; Bafna, V.; Ideker, T. Inferring gene ontologies from pairwise similarity data. Bioinformatics 2014, 30, i34–i42. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Islam, A.B.M.M.K.; Khan, M.A.-A.-K.; Ahmed, R.; Hossain, M.S.; Kabir, S.M.T.; Islam, M.S.; Siddiki, A.M.A.M.Z. Transcriptome of nasopharyngeal samples from COVID-19 patients and a comparative analysis with other SARS-CoV-2 infection models reveal disparate host responses against SARS-CoV-2. J. Transl. Med. 2021, 19, 32. [Google Scholar] [CrossRef]
- Sigrist, C.J.; Bridge, A.; le Mercier, P. A potential role for integrins in host cell entry by SARS-CoV-2. Antiviral Res. 2020, 177, 104759. [Google Scholar] [CrossRef]
- EA, J.A.; Jones, I.M. Membrane binding proteins of coronaviruses. Future Virol. 2019, 14, 275–286. [Google Scholar]
- Giri, R.; Bhardwaj, T.; Shegane, M.; Gehi, B.R.; Kumar, P.; Gadhave, K.; Oldfield, C.J.; Uversky, V.N. Understanding COVID-19 via comparative analysis of dark proteomes of SARS-CoV-2, human SARS and bat SARS-like coronaviruses. Cell Mol. Life Sci. 2021, 78, 1655–1688. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Shimakawa, G.; Tamura, M.; Yokosawa, H.; Arata, Y. ISG15 regulates RANKL-induced osteoclastogenic differentiation of RAW264 cells. Biol. Pharm. Bull. 2015, 38, 482–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokado, M.; Okada, Y.; Goto, M.; Ishikawa, K.; Miyamoto, T.; Yamanaka, O.; Fujiwara, S.; Saika, S. Increased fragility, impaired differentiation, and acceleration of migration of corneal epithelium of epiplakin-null mice. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3780–3789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Peng, F.; Liu, Z.; Jiang, F.; Li, L.; Gao, S.; Wang, G.; Song, J.; Ruan, E.; Shao, Z.; et al. CYR61/CCN1 stimulates proliferation and differentiation of osteoblasts in vitro and contributes to bone remodeling in vivo in myeloma bone disease. Int. J. Oncol. 2017, 50, 631–639. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Gao, Y.; Qin, J.; Kuang, C.-Y.; Song, M.-B.; Yu, S.-Y.; Cui, B.; Chen, J.-F.; Huang, L. CCN1 promotes the differentiation of endothelial progenitor cells and reendothelialization in the early phase after vascular injury. Basic Res. Cardiol. 2010, 105, 713–724. [Google Scholar] [CrossRef]
- Geng, A.; Qiu, R.; Murai, K.; Liu, J.; Wu, X.; Zhang, H.; Farhoodi, H.; Duong, N.; Jiang, M.; Yee, J.-K.; et al. KIF20A/MKLP2 regulates the division modes of neural progenitor cells during cortical development. Nat. Commun. 2018, 9, 2707. [Google Scholar] [CrossRef] [Green Version]
- Wada, M.; Lokugamage, K.G.; Nakagawa, K.; Narayanan, K.; Makino, S. Interplay between coronavirus, a cytoplasmic RNA virus, and nonsense-mediated mRNA decay pathway. Proc. Natl. Acad. Sci. USA 2018, 115, E10157–E10166. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Pluhackova, K.; Böckmann, R.A. The Multifaceted Role of SNARE Proteins in Membrane Fusion. Front. Physiol. 2017, 8, 5. [Google Scholar] [CrossRef] [Green Version]
- Abu-Farha, M.; Thanaraj, T.A.; Qaddoumi, M.G.; Hashem, A.; Abubaker, J.; Al-Mulla, F. The Role of Lipid Metabolism in COVID-19 Virus Infection and as a Drug Target. Int. J. Mol. Sci. 2020, 21, 3544. [Google Scholar] [CrossRef]
- Tiku, V.; Tan, M.-W.; Dikic, I. Mitochondrial Functions in Infection and Immunity. Trends Cell Biol. 2020, 30, 263–275. [Google Scholar] [CrossRef] [Green Version]
- Aguirre, S.; Luthra, P.; Sanchez-Aparicio, M.T.; Maestre, A.M.; Patel, J.; Lamothe, F.; Fredericks, A.C.; Tripathi, S.; Zhu, T.; Pintado-Silva, J.; et al. Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nat. Microbiol. 2017, 2, 17037. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Xie, W.; Liu, M. Alteration of cell junctions during viral infection. Thorac. Cancer 2020, 11, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Roy, U.; Barber, P.; Tse-Dinh, Y.-C.; Batrakova, E.V.; Mondal, D.; Nair, M. Role of MRP transporters in regulating antimicrobial drug inefficacy and oxidative stress-induced pathogenesis during HIV-1 and TB infections. Front. Microbiol. 2015, 6, 948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortese, M.; Lee, J.; Cerikan, B.; Neufeldt, C.J.; Oorschot, V.M.J.; Köhrer, S.; Hennies, J.; Schieber, N.L.; Ronchi, P.; Mizzon, G.; et al. Integrative Imaging Reveals SARS-CoV-2-Induced Reshaping of Subcellular Morphologies. Cell Host Microbe 2020, 28, 853–866.e5. [Google Scholar] [CrossRef] [PubMed]
- York, A. Exploiting peroxisomes. Nat. Rev. Microbiol. 2018, 16, 659. [Google Scholar] [CrossRef]
- Bianchi, M.; Borsetti, A.; Ciccozzi, M.; Pascarella, S. SARS-Cov-2 ORF3a: Mutability and function. Int. J. Biol. Macromol. 2021, 170, 820–826. [Google Scholar] [CrossRef]
- Wang, X.; Melino, G.; Shi, Y. Actively or passively deacidified lysosomes push beta-coronavirus egress. Cell Death Dis. 2021, 12, 235. [Google Scholar] [CrossRef]
- Martin-Serrano, J.; Eastman, S.W.; Chung, W.; Bieniasz, P.D. HECT ubiquitin ligases link viral and cellular PPXY motifs to the vacuolar protein-sorting pathway. J. Cell Biol. 2005, 168, 89–101. [Google Scholar] [CrossRef]
- Lee, J.; Huang, W.; Lee, H.; van de Leemput, J.; Kane, M.A.; Han, Z. Characterization of SARS-CoV-2 proteins reveals Orf6 pathogenicity, subcellular localization, host interactions and attenuation by Selinexor. Cell Biosci. 2021, 11, 58. [Google Scholar] [CrossRef]
- Kuo, M.-L.; Lee, M.B.-E.; Tang, M.; Besten, W.D.; Hu, S.; Sweredoski, M.J.; Hess, S.; Chou, C.-M.; Changou, C.A.; Su, M.; et al. PYCR1 and PYCR2 Interact and Collaborate with RRM2B to Protect Cells from Overt Oxidative Stress. Sci. Rep. 2016, 6, 18846. [Google Scholar] [CrossRef]
- Bagga, S.; Bouchard, M.J. Cell cycle regulation during viral infection. Methods Mol. Biol. 2014, 1170, 165–227. [Google Scholar] [PubMed]
- Zhang, H.; Zheng, H.; Zhu, J.; Dong, Q.; Wang, J.; Fan, H.; Chen, Y.; Zhang, X.; Han, X.; Li, Q.; et al. Ubiquitin-Modified Proteome of SARS-CoV-2-Infected Host Cells Reveals Insights into Virus-Host Interaction and Pathogenesis. J. Proteome Res. 2021, 20, 2224–2239. [Google Scholar] [CrossRef] [PubMed]
- Flower, T.G.; Buffalo, C.Z.; Hooy, R.M.; Allaire, M.; Ren, X.; Hurley, J.H. Structure of SARS-CoV-2 ORF8, a rapidly evolving immune evasion protein. Proc. Natl. Acad. Sci. USA 2021, 118, e2021785118. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Y.; Li, Y.; Huang, F.; Luo, B.; Yuan, Y.; Xia, B.; Ma, X.; Yang, T.; Yu, F.; et al. The ORF8 protein of SARS-CoV-2 mediates immune evasion through down-regulating MHC-Iota. Proc. Natl. Acad. Sci. USA 2021, 118, e2024202118. [Google Scholar] [CrossRef]
- Young, B.E.; Fong, S.; Chan, Y.; Mak, T.; Ang, L.W.; Anderson, D.E.; Lee, C.Y.; Amrun, S.N.; Lee, B.; Goh, Y.S.; et al. Effects of a major deletion in the SARS-CoV-2 genome on the severity of infection and the inflammatory response: An observational cohort study. Lancet 2020, 396, 603–611. [Google Scholar] [CrossRef]
- Vigerust, D.J.; Shepherd, V.L. Virus glycosylation: Role in virulence and immune interactions. Trends Microbiol. 2007, 15, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Bowden, T.A.; Wilson, I.A.; Crispin, M. Exploitation of glycosylation in enveloped virus pathobiology. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1480–1497. [Google Scholar] [CrossRef]
- Kim, S.-A.; Taylor, G.S.; Torgersen, K.M.; Dixon, J.E. Myotubularin and MTMR2, phosphatidylinositol 3-phosphatases mutated in myotubular myopathy and type 4B Charcot-Marie-Tooth disease. J. Biol. Chem. 2002, 277, 4526–4531. [Google Scholar] [CrossRef] [Green Version]
- Angelini, M.M.; Akhlaghpour, M.; Neuman, B.W.; Buchmeier, M.J. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio 2013, 4, e00524-13. [Google Scholar] [CrossRef] [Green Version]
- Byun, H.; Gou, Y.; Zook, A.; Lozano, M.M.; Dudley, J.P. ERAD and how viruses exploit it. Front. Microbiol. 2014, 5, 330. [Google Scholar] [CrossRef] [Green Version]
- Aly, H.H.; Suzuki, J.; Watashi, K.; Chayama, K.; Hoshino, S.; Hijikata, M.; Kato, T.; Wakita, T. RNA Exosome Complex Regulates Stability of the Hepatitis B Virus X-mRNA Transcript in a Non-stop-mediated (NSD) RNA Quality Control Mechanism. J. Biol. Chem. 2016, 291, 15958–15974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyashita, M.; Oshiumi, H.; Matsumoto, M.; Seya, T. DDX60, a DEXD/H box helicase, is a novel antiviral factor promoting RIG-I-like receptor-mediated signaling. Mol. Cell Biol. 2011, 31, 3802–3819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnapp, L.M.; Donohoe, S.; Chen, J.; Sunde, D.A.; Kelly, P.M.; Ruzinski, J.; Martin, T.; Goodlett, D.R. Mining the acute respiratory distress syndrome proteome: Identification of the insulin-like growth factor (IGF)/IGF-binding protein-3 pathway in acute lung injury. Am. J. Pathol. 2006, 169, 86–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahasic, A.M.; Zhai, R.; Su, L.; Zhao, Y.; Aronis, K.N.; Thompson, B.T.; Mantzoros, C.S.; Christiani, D.C. IGF1 and IGFBP3 in acute respiratory distress syndrome. Eur. J. Endocrinol. 2012, 166, 121–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Sancho, L.; Lewinski, M.K.; Pache, L.; Stoneham, C.A.; Yin, X.; Becker, M.E.; Pratt, D.; Churas, C.; Rosenthal, S.B.; Liu, S.; et al. Functional landscape of SARS-CoV-2 cellular restriction. Mol. Cell 2021, 81, 2656–2668.e8. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.L. Stress Granules and Virus Replication. Future Virol. 2011, 6, 1329–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cascarina, S.M.; Ross, E.D. A proposed role for the SARS-CoV-2 nucleocapsid protein in the formation and regulation of biomolecular condensates. FASEB J. 2020, 34, 9832–9842. [Google Scholar] [CrossRef]
- Wang, J.; Shi, C.; Xu, Q.; Yin, H. SARS-CoV-2 nucleocapsid protein undergoes liquid-liquid phase separation into stress granules through its N-terminal intrinsically disordered region. Cell Discov. 2021, 7, 5. [Google Scholar] [CrossRef]
- Lu, F. SARS-CoV-2 ORF9c: A mysterious membrane-anchored protein that regulates immune evasion? Nat. Rev. Immunol. 2020, 20, 648. [Google Scholar] [CrossRef]
- Shi, C.-S.; Nabar, N.R.; Huang, N.; Kehrl, J.H. SARS-Coronavirus Open Reading Frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 2019, 5, 101. [Google Scholar] [CrossRef] [Green Version]
- Samson, A.L.; Zhang, Y.; Geoghegan, N.D.; Gavin, X.J.; Davies, K.A.; Mlodzianoski, M.J.; Whitehead, L.W.; Frank, D.; Garnish, S.E.; FitzGibbon, C.; et al. MLKL trafficking and accumulation at the plasma membrane control the kinetics and threshold for necroptosis. Nat. Commun. 2020, 11, 3151. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.K.W.; Hui, K.P.Y.; Gu, H.; Ko, R.L.W.; Krishnan, P.; Ng, D.Y.M.; Liu, G.Y.Z.; Wan, C.K.C.; Cheung, M.; Ng, K.-C.; et al. Introduction of ORF3a-Q57H SARS-CoV-2 Variant Causing Fourth Epidemic Wave of COVID-19, Hong Kong, China. Emerg. Infect. Dis. 2021, 27, 1492–1495. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, P.; Niyogi, S. ORF3a mutation associated with higher mortality rate in SARS-CoV-2 infection. Epidemiol. Infect. 2020, 148, e262. [Google Scholar] [CrossRef] [PubMed]
- Corsello, S.; Bittker, J.A.; Liu, Z.; Gould, J.; McCarren, P.; Hirschman, J.E.; Johnston, S.E.; Vrcic, A.; Wong, B.; Khan, M.; et al. The Drug Repurposing Hub: A next-generation drug library and information resource. Nat. Med. 2017, 23, 405–408. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Wang, F.; Tang, J.; Nussinov, R.; Cheng, F. Artificial intelligence in COVID-19 drug repurposing. Lancet Digit. Heal. 2020, 2, e667–e676. [Google Scholar] [CrossRef]
- Riva, L.; Yuan, S.; Yin, X.; Martin-Sancho, L.; Matsunaga, N.; Pache, L.; Burgstaller-Muehlbacher, S.; de Jesus, P.D.; Teriete, P.; Hull, M.V.; et al. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 2020, 586, 113–119. [Google Scholar] [CrossRef]
- Cho, J.; Lee, Y.J.; Kim, J.H.; Kim, S.I.; Kim, S.S.; Choi, B.; Choi, J.-H. Antiviral activity of digoxin and ouabain against SARS-CoV-2 infection and its implication for COVID-19. Sci. Rep. 2020, 10, 16200. [Google Scholar] [CrossRef]
- Kamitani, W.; Huang, C.; Narayanan, K.; Lokugamage, K.G.; Makino, S. A two-pronged strategy to suppress host protein synthesis by SARS coronavirus Nsp1 protein. Nat. Struct. Mol. Biol. 2009, 16, 1134–1140. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.; Xu, W.; Liu, S.-W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Nelson, C.W.; Ardern, Z.; Goldberg, T.L.; Meng, C.; Kuo, C.; Ludwig, C.; Kolokotronis, S.; Wei, X.; Biodiversity Research Center, Academia Sinica, Taiwan; Institute for Comparative Genomics, American Museum of Natural History, United States; et al. Dynamically evolving novel overlapping gene as a factor in the SARS-CoV-2 pandemic. Elife 2020, 9, e59633. [Google Scholar] [CrossRef]
- Edwards, D.A.; Ausiello, D.; Salzman, J.; Devlin, T.; Langer, R.; Beddingfield, B.J.; Fears, A.C.; Doyle-Meyers, L.A.; Redmann, R.K.; Killeen, S.Z.; et al. Exhaled aerosol increases with COVID-19 infection, age, and obesity. Proc. Natl. Acad. Sci. USA 2021, 118, e2021830118. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Liu, X.; Wang, T.; Liu, F.; Zhu, A.; Lin, Y.; Luo, J.; Ye, F.; He, J.; Zhao, J.; et al. Elevated MUC1 and MUC5AC mucin protein levels in airway mucus of critical ill COVID-19 patients. J. Med. Virol. 2021, 93, 582–584. [Google Scholar] [CrossRef] [PubMed]
- Elena, S.F.; Carrasco, P.; Daròs, J.-A.; Sanjuán, R. Mechanisms of genetic robustness in RNA viruses. EMBO Rep. 2006, 7, 168–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krakauer, D.C.; Plotkin, J.B. Redundancy, antiredundancy, and the robustness of genomes. Proc. Natl. Acad. Sci. USA 2002, 99, 1405–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
May, D.G.; Martin-Sancho, L.; Anschau, V.; Liu, S.; Chrisopulos, R.J.; Scott, K.L.; Halfmann, C.T.; Díaz Peña, R.; Pratt, D.; Campos, A.R.; et al. A BioID-Derived Proximity Interactome for SARS-CoV-2 Proteins. Viruses 2022, 14, 611. https://doi.org/10.3390/v14030611
May DG, Martin-Sancho L, Anschau V, Liu S, Chrisopulos RJ, Scott KL, Halfmann CT, Díaz Peña R, Pratt D, Campos AR, et al. A BioID-Derived Proximity Interactome for SARS-CoV-2 Proteins. Viruses. 2022; 14(3):611. https://doi.org/10.3390/v14030611
Chicago/Turabian StyleMay, Danielle G., Laura Martin-Sancho, Valesca Anschau, Sophie Liu, Rachel J. Chrisopulos, Kelsey L. Scott, Charles T. Halfmann, Ramon Díaz Peña, Dexter Pratt, Alexandre R. Campos, and et al. 2022. "A BioID-Derived Proximity Interactome for SARS-CoV-2 Proteins" Viruses 14, no. 3: 611. https://doi.org/10.3390/v14030611
APA StyleMay, D. G., Martin-Sancho, L., Anschau, V., Liu, S., Chrisopulos, R. J., Scott, K. L., Halfmann, C. T., Díaz Peña, R., Pratt, D., Campos, A. R., & Roux, K. J. (2022). A BioID-Derived Proximity Interactome for SARS-CoV-2 Proteins. Viruses, 14(3), 611. https://doi.org/10.3390/v14030611