Early Emergence Phase of SARS-CoV-2 Delta Variant in Florida, US

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset
2.2. Phylodynamic Analysis
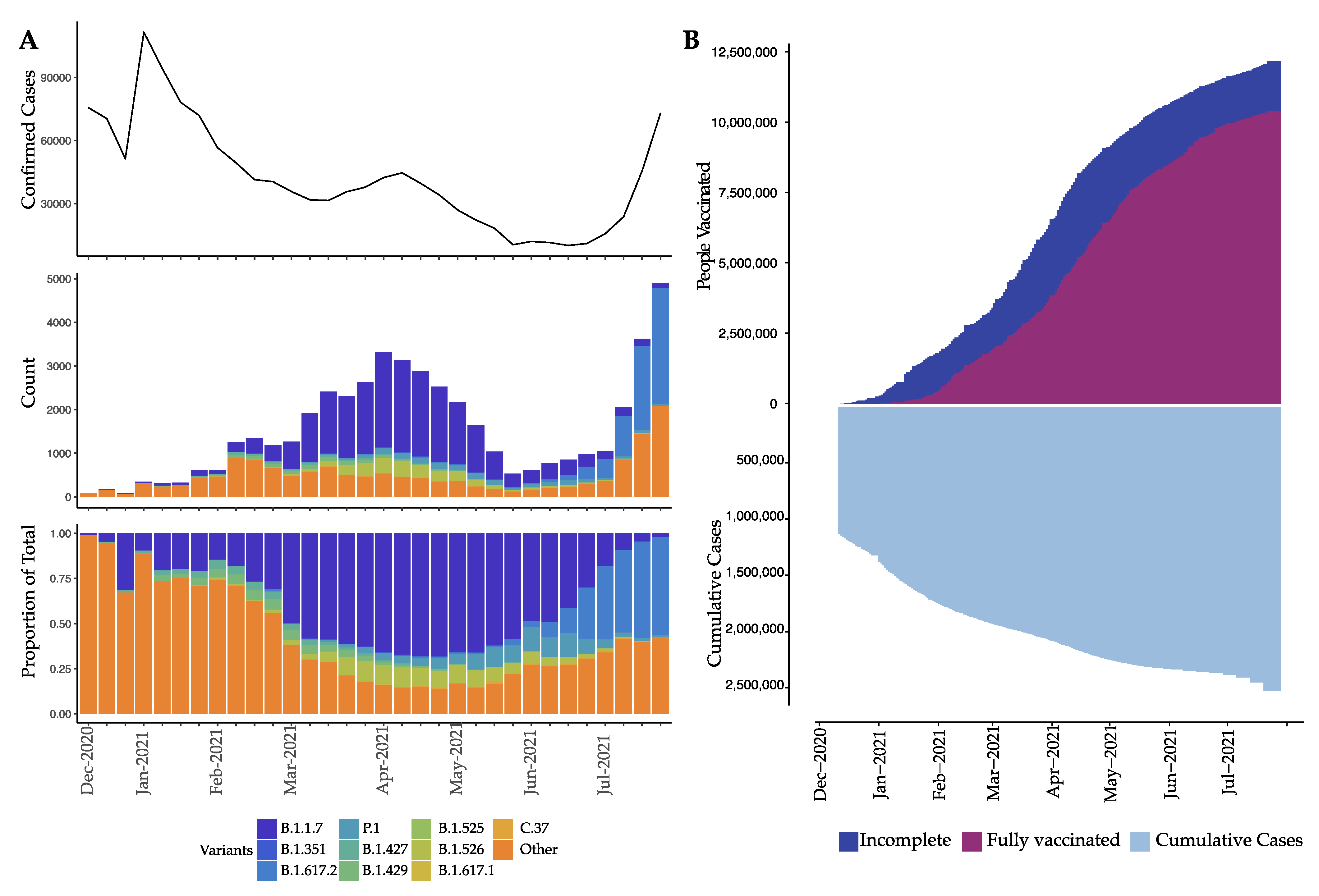
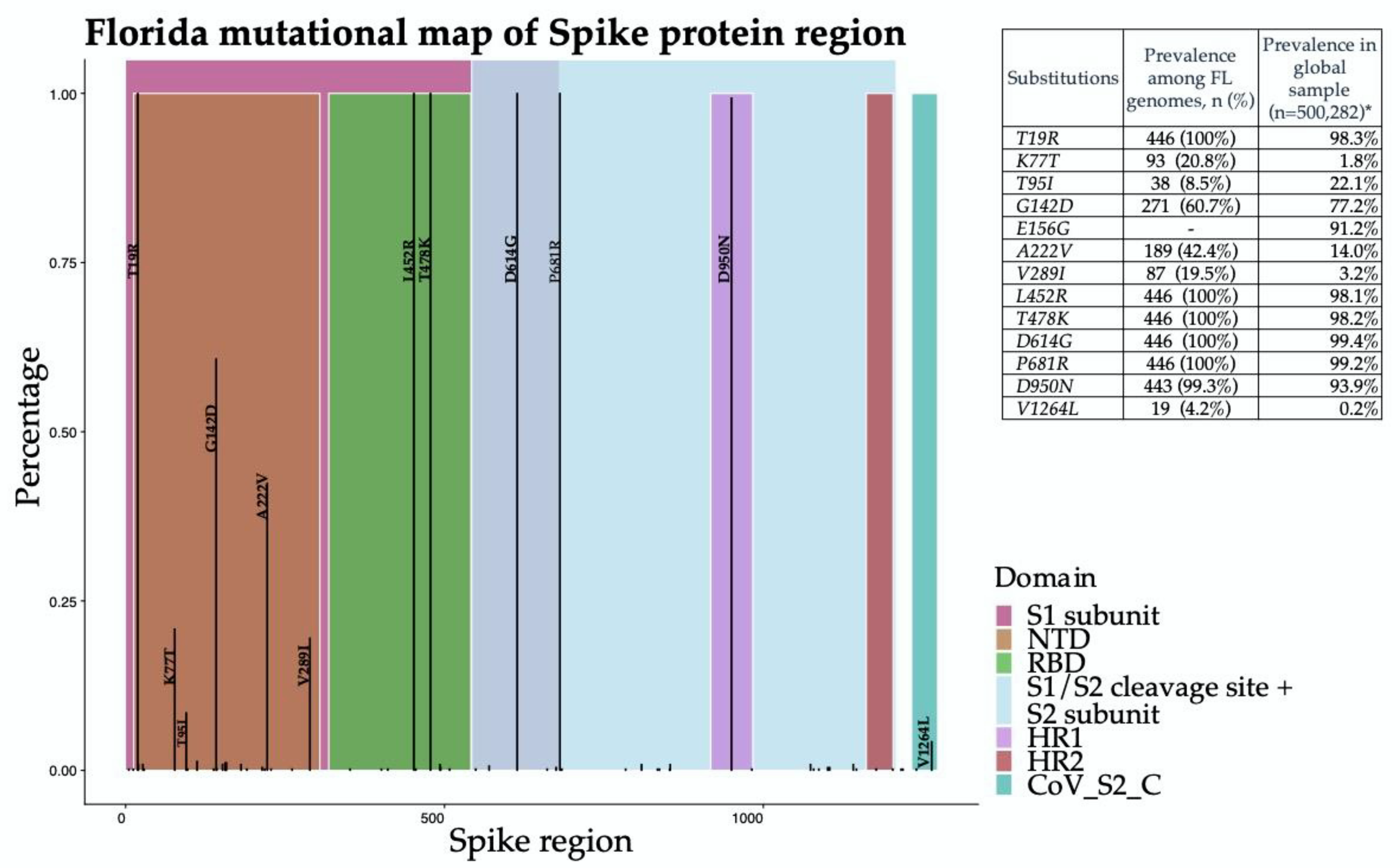
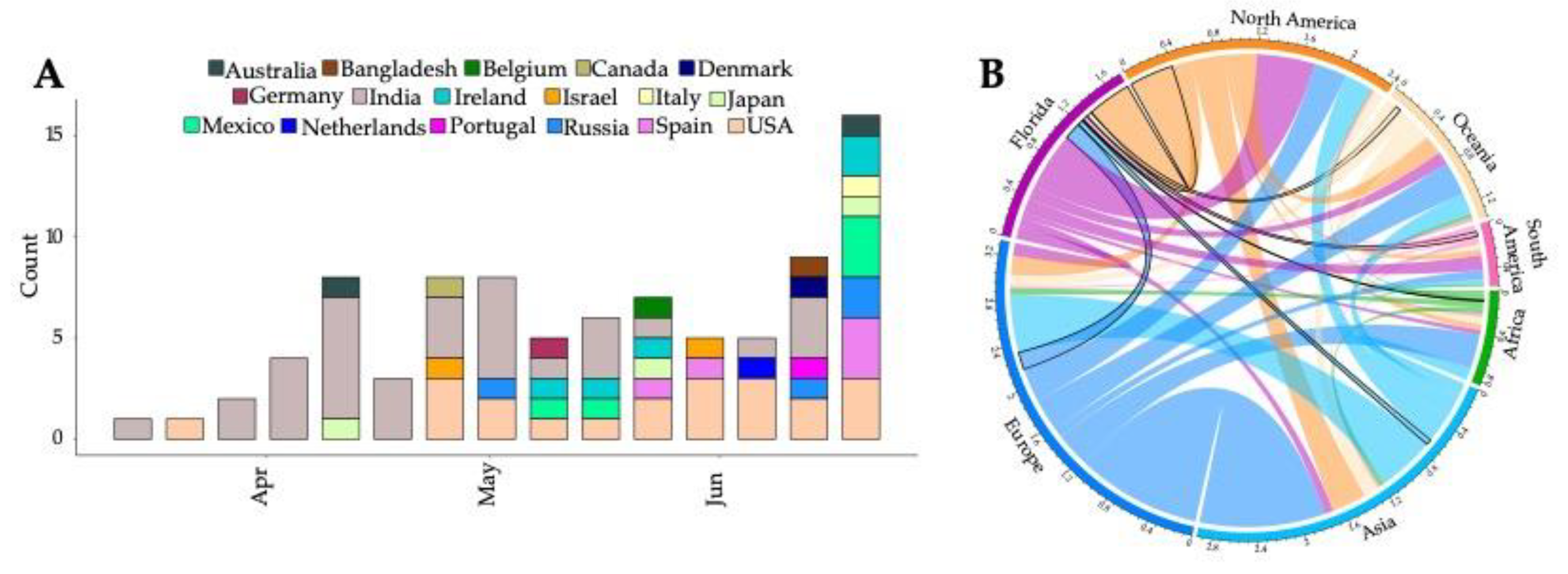
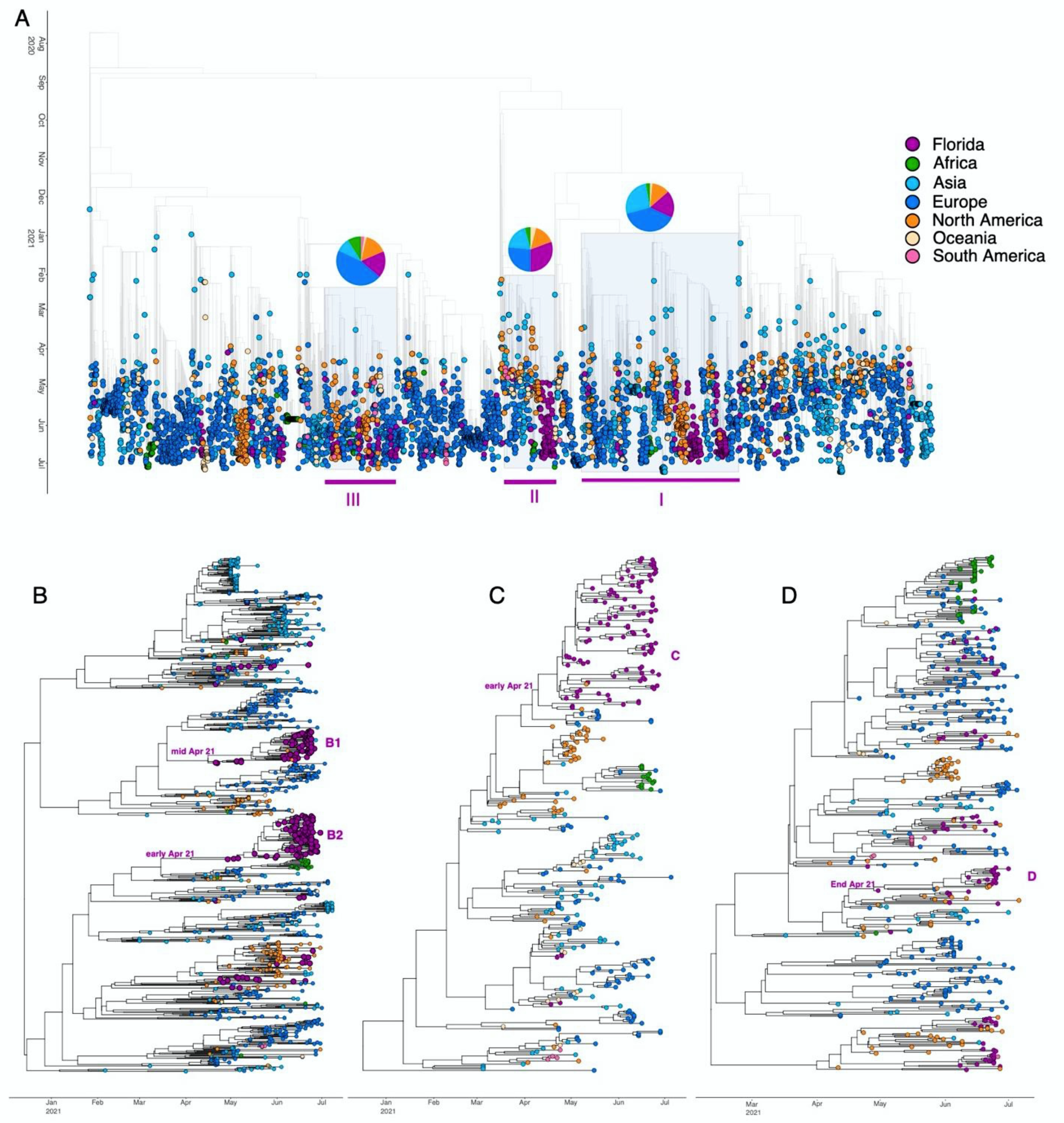
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johns Hopkins. COVID-19 Map. Johns Hopkins Coronavirus Resource Center. Available online: https://coronavirus.jhu.edu/ (accessed on 26 July 2021).
- Abdool Karim, S.S.; de Oliveira, T. New SARS-CoV-2 Variants—Clinical, Public Health, and Vaccine Implications. N. Engl. J. Med. 2021, 384, 1866–1868. [Google Scholar] [CrossRef] [PubMed]
- Thye, A.Y.K.; Law, J.W.F.; Pusparajah, P.; Letchumanan, V.; Chan, K.G.; Lee, L.H. Emerging SARS-CoV-2 Variants of Concern (VOCs): An Impending Global Crisis. Biomedicines 2021, 9, 1303. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G Variant Exhibits Efficient Replication Ex Vivo and Transmission in vivo. Science 2020, 370, 1464–1468. [Google Scholar] [CrossRef] [PubMed]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; et al. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell 2020, 183, 739–751.e8. [Google Scholar] [CrossRef]
- Elbe, S.; Buckland-Merrett, G. Data, Disease and Diplomacy: GISAID’s Innovative Contribution to Global Health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [Green Version]
- CDC. SARS-CoV-2 Variant Classifications and Definitions; CDC: Atlanta, GA, US, 2021. Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-classifications.html (accessed on 26 July 2021).
- World Health Organization. Tracking SARS-CoV-2 Variants. 2021. Available online: https://www.who.int/en/activities/tracking-SARS-Co (accessed on 26 July 2021).
- Robishaw, J.D.; Alter, S.M.; Solano, J.J.; Shih, R.D.; DeMets, D.L.; Maki, D.G.; Hennekens, C.H. Genomic Surveillance to Combat COVID-19: Challenges and Opportunities. Lancet Microbe 2021, 2, e481–e484. [Google Scholar] [CrossRef]
- Torjesen, I. Covid-19: Delta Variant Is Now UK’s Most Dominant Strain and Spreading through Schools. BMJ 2021, 373, n1445. [Google Scholar] [CrossRef]
- Bolze, A.; Luo, S.; White, S.; Cirulli, E.T.; Wyman, D.; Dei Rossi, A.; Machado, H.; Cassens, T.; Jacobs, S.; Barrett, K.M.; et al. SARS-CoV-2 variant Delta rapidly displaced variant Alpha in the United States and led to higher viral loads. Cell Rep. Med. 2022, 3, 100564. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Althaus, C.L.; Giovanetti, M.; San, J.E.; Giandhari, J.; Pillay, S.; Naidoo, Y.; Ramphal, U.; Msomi, N.; et al. Rapid Replacement of the Beta Variant by the Delta Variant in South Africa. medRxiv 2021. [Google Scholar] [CrossRef]
- Sheikh, A.; McMenamin, J.; Taylor, B.; Robertson, C. SARS-CoV-2 Delta VOC in Scotland: Demographics, Risk of Hospital Admission, and Vaccine Effectiveness. Lancet 2021, 397, 2461–2462. [Google Scholar] [CrossRef]
- U.S. CDC Delta Variant: What We Know about the Science. Available online: https://stacks.cdc.gov/view/cdc/108671 (accessed on 21 August 2021).
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence That D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. medRxiv 2020, 184, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Daniloski, Z.; Jordan, T.X.; Ilmain, J.K.; Guo, X.; Bhabha, G.; Sanjana, N.E. The Spike D614G Mutation Increases SARS-CoV-2 Infection of Multiple Human Cell Types. eLife 2021, 10, e65365. [Google Scholar] [CrossRef]
- Deng, X.; Garcia-Knight, M.A.; Khalid, M.M.; Servellita, V.; Wang, C.; Morris, M.K.; Sotomayor-González, A.; Glasner, D.R.; Reyes, K.R.; Gliwa, A.S.; et al. Transmission, Infectivity, and Neutralization of a Spike L452R SARS-CoV-2 Variant. Cell 2021, 184, 3426–3437.e8. [Google Scholar] [CrossRef]
- Allen, H.; Vusirikala, A.; Flannagan, J.; Twohig, K.A.; Zaidi, A.; Chudasama, D.; Lamagni, T.; Groves, N.; Turner, C.; Rawlinson, C.; et al. Household transmission of COVID-19 cases associated with SARS-CoV-2 delta variant (B. 1.617. 2): National case-control study. Lancet Reg. Health-Eur. 2022, 12, 100252. [Google Scholar] [CrossRef]
- Liu, C.; Ginn, H.M.; Dejnirattisai, W.; Ren, J.; Stuart, D.I.; Screaton, G.R. Article Reduced Neutralization of SARS-CoV-2 B. 1. 617 by Vaccine and Convalescent Serum Reduced Neutralization of SARS-CoV-2 B.1.617 by Vaccine and Convalescent Serum. Cell 2021, 184, 1–17. [Google Scholar] [CrossRef]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced Sensitivity of Infectious SARS-CoV-2 Variant B.1.617.2 to Monoclonal Antibodies and Sera from Convalescent and Vaccinated Individuals. bioRxiv 2021. [Google Scholar] [CrossRef]
- Yadav, P.D.; Sapkal, G.N.; Ella, R.; Sahay, R.R.; Nyayanit, D.A.; Patil, D.Y.; Deshpande, G.; Shete, A.M.; Gupta, N.; Mohan, V.K.; et al. Neutralization against B. 1.351 and B. 1.617. 2 with sera of COVID-19 recovered cases and vaccinees of BBV152. BioRxiv 2021. [Google Scholar] [CrossRef]
- Wall, E.C.; Wu, M.; Harvey, R.; Kelly, G.; Warchal, S.; Sawyer, C.; Daniels, R.; Adams, L.; Hobson, P.; Hatipoglu, E.; et al. AZD1222-Induced Neutralising Antibody Activity against SARS-CoV-2 Delta VOC. Lancet 2021, 398, 207–209. [Google Scholar] [CrossRef]
- Challen, R.; Dyson, L.; Overton, C.E.; Guzman-Rincon, L.M.; Hill, E.M.; Stage, H.B.; Brooks-Pollock, E.; Pellis, L.; Scarabel, F.; Pascall, D.J.; et al. Early epidemiological signatures of novel SARS-CoV-2 variants: Establishment of B. 1.617. 2 in England. Medrxiv. 2021. [Google Scholar] [CrossRef]
- Elliott, P.; Haw, D.; Wang, H.; Eales, O.; Walters, C.E.; Ainslie, K.E.C.; Atchison, C.; Fronterre, C.; Diggle, P.J.; Page, A.J.; et al. Exponential Growth, High Prevalence of SARS-CoV-2, and Vaccine Effectiveness Associated with the Delta Variant. Science 2021, 374, eabl9551. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.; Lim, J.-S.; Song, S.-A.; Achangwa, C.; Sim, W.; Kim, G.; Ryu, S. Transmission Dynamics of the Delta Variant of SARS-CoV-2 Infections in South Korea. J. Infect. Dis. 2021, 5, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Likos, A.; Griffin, I.; Bingham, A.M.; Stanek, D.; Fischer, M.; White, S.; Hamilton, J.; Eisenstein, L.; Atrubin, D.; Mulay, P.; et al. Local Mosquito-Borne Transmission of Zika Virus—Miami-Dade and Broward Counties, Florida, June–August 2016. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 1032–1038. [Google Scholar] [CrossRef] [Green Version]
- Singanayagam, A.; Hakki, S.; Dunning, J.; Madon, K.J.; Crone, M.A.; Koycheva, A.; Derqui-Fernandez, N.; Barnett, J.L.; Whitfield, M.G.; Varro, R.; et al. Community Transmission and Viral Load Kinetics of the SARS-CoV-2 Delta (B.1.617.2) Variant in Vaccinated and Unvaccinated Individuals in the UK: A Prospective, Longitudinal, Cohort Study. Lancet Infect. Dis. 2022, 22, 183–195. [Google Scholar] [CrossRef]
- Mahase, E. Delta Variant: What Is Happening with Transmission, Hospital Admissions, and Restrictions? BMJ 2021, 373, n1513. [Google Scholar] [CrossRef]
- Del Rio, C.; Malani, P.N.; Omer, S.B. Confronting the Delta Variant of SARS-CoV-2, Summer 2021. JAMA 2021, 326, 1001–1002. [Google Scholar] [CrossRef]
- Marini, S.; Mavian, C.; Riva, A.; Prosperi, M.; Salemi, M.; Rife Magalis, B. Optimizing Viral Genome Subsampling by Genetic Diversity and Temporal Distribution (TARDiS) for Phylogenetics. Bioinformatics 2022, 38, 856–860. [Google Scholar] [CrossRef]
- Moshiri, N. ViralMSA: Massively Scalable Reference-Guided Multiple Sequence Alignment of Viral Genomes. Bioinformatics 2021, 37, 714–716. [Google Scholar] [CrossRef]
- Larsson, A. AliView: A Fast and Lightweight Alignment Viewer and Editor for Large Datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Lam, T.T.; Carvalho, L.M.; Pybus, O.G. Exploring the Temporal Structure of Heterochronous Sequences Using TempEst (Formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volz, E.M.; Frost, S.D.W. Scalable Relaxed Clock Phylogenetic Dating. Virus Evol. 2017, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Giovanetti, M.; Cella, E.; Benedetti, F.; Magalis, B.R.; Fonseca, V.; Fabris, S.; Campisi, G.; Ciccozzi, A.; Angeletti, S.; Borsetti, A.; et al. SARS-CoV-2 Shifting Transmission Dynamics and Hidden Reservoirs Limited the Efficacy of Public Health Interventions in Italy. Commun. Biol. 2021. [Google Scholar] [CrossRef]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-Likelihood Phylodynamic Analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, M.; Kishino, H.; Yano, T. aki Dating of the Human-Ape Splitting by a Molecular Clock of Mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef]
- Griffiths, R.C.; Tavaré, S. Sampling Theory for Neutral Alleles in a Varying Environment. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1994, 344, 403–410. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Oster, A.M.; France, A.M.; Panneer, N.; Cheryl Bañez Ocfemia, M.; Campbell, E.; Dasgupta, S.; Switzer, W.M.; Wertheim, J.O.; Hernandez, A.L. Identifying Clusters of Recent and Rapid HIV Transmission through Analysis of Molecular Surveillance Data. J. Acquir. Immune Defic. Syndr. 2018, 79, 543–550. [Google Scholar] [CrossRef]
- Rich, S.N.; Richards, V.L.; Mavian, C.N.; Switzer, W.M.; Magalis, B.R.; Poschman, K.; Geary, S.; Broadway, S.E.; Bennett, S.B.; Blanton, J.; et al. Employing Molecular Phylodynamic Methods to Identify and Forecast HIV Transmission Clusters in Public Health Settings: A Qualitative Study. Viruses 2020, 12, 921. [Google Scholar] [CrossRef] [PubMed]
- Ballotpedia Documenting Minnesota’s Path to Recovery from the Coronavirus (COVID-19) Pandemic, 2020–2021. Available online: https://ballotpedia.org/Documenting_Florida%27s_path_to_recovery_from_the_coronavirus_(COVID-19)_pandemic,_2020-2021 (accessed on 21 December 2021).
- Florida Department of Health. Latest Vaccine Updates-Florida Department of Health COVID-19 Outbreak. Available online: https://floridahealthcovid19.gov/ (accessed on 27 July 2021).
- Mavian, C.; Marini, S.; Prosperi, M.; Salemi, M. A Snapshot of SARS-CoV-2 Genome Availability up to 30th March, 2020 and Its Implications. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Mavian, C.; Marini, S.; Manes, C.; Capua, I.; Prosperi, M.; Salemi, M. Regaining Perspective on SARS-CoV-2 Molecular Tracing and Its Implications. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Xavier, J.; Giovanetti, M.; Adelino, T.; Fonseca, V.; Barbosa da Costa, A.V.; Ribeiro, A.A.; Felicio, K.N.; Duarte, C.G.; Ferreira Silva, M.V.; Salgado, Á.; et al. The Ongoing COVID-19 Epidemic in Minas Gerais, Brazil: Insights from Epidemiological Data and SARS-CoV-2 Whole Genome Sequencing. Emerg. Microbes Infect. 2020, 9, 1824–1834. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. Phylodynamic Analysis|176 Genomes|6 Mar 2020–Novel 2019 Coronavirus/NCoV-2019 Genomic Epidemiology-Virological. Available online: https://virological.org/t/phylodynamic-analysis-176-genomes-6-mar-2020/356 (accessed on 7 April 2021).
- Mathieu, E.; Ritchie, H.; Ortiz-Ospina, E.; Roser, M.; Hasell, J.; Appel, C.; Giattino, C.; Rodés-Guirao, L. A Global Database of COVID-19 Vaccinations. Nat. Hum. Behav. 2021, 5, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Alpert, T.; Lasek-Nesselquist, E.; Brito, A.F.; Valesano, A.L. Early Introductions and Community Transmission of SARS-CoV-2 Variant B. 1.1. 7 in the United States. medRxiv 2021, 184, 2595–2604. [Google Scholar]
- Washington, N.L.; Gangavarapu, K.; Zeller, M.; Bolze, A.; Cirulli, E.T.; Schiabor Barrett, K.M.; Larsen, B.B.; Anderson, C.; White, S.; Cassens, T.; et al. Emergence and Rapid Transmission of SARS-CoV-2 B.1.1.7 in the United States. Cell 2021, 184, 2587–2594.e7. [Google Scholar] [CrossRef]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid Epidemic Expansion of the SARS-CoV-2 Omicron Variant in Southern Africa. Nature 2022, 1–10. [Google Scholar] [CrossRef]
- Sallam, M.; Al-Sanafi, M.; Sallam, M. A Global Map of COVID-19 Vaccine Acceptance Rates per Country: An Updated Concise Narrative Review. J. Multidiscip. Healthc. 2022, 15, 21. [Google Scholar] [CrossRef]
- Ramuta, M.D.; Newman, C.M.; Brakefield, S.F.; Stauss, M.R.; Wiseman, R.W.; Kita-Yarbro, A.; O’Connor, E.J.; Dahal, N.; Lim, A.; Poulsen, K.P.; et al. SARS-CoV-2 and other respiratory pathogens are detected in continuous air samples from congregate settings. medRxiv 2022. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subset/Subtree | N of Taxa | % of FL Taxa | Minor Mutations Associated | Oster Value |
|---|---|---|---|---|
| Subset I | 1026 | 18.3% | A222V, V289I | 1.31 |
| Subtree B1 | 74 | 95.9% | A222V | 1.59 |
| Subtree B2 | 88 | 100.0% | A222V, V289I | 1.83 |
| Subset II | 326 | 30.7% | K77T | 1.52 |
| Subtree C | 91 | 98.9% | K77T | 1.51 |
| Subset III | 470 | 17.7% | - | 1.45 |
| Subtree D | 22 | 81.8% | - | 2.14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cella, E.; Ali, S.; Schmedes, S.E.; Rife Magalis, B.; Marini, S.; Salemi, M.; Blanton, J.; Azarian, T. Early Emergence Phase of SARS-CoV-2 Delta Variant in Florida, US. Viruses 2022, 14, 766. https://doi.org/10.3390/v14040766
Cella E, Ali S, Schmedes SE, Rife Magalis B, Marini S, Salemi M, Blanton J, Azarian T. Early Emergence Phase of SARS-CoV-2 Delta Variant in Florida, US. Viruses. 2022; 14(4):766. https://doi.org/10.3390/v14040766
Chicago/Turabian StyleCella, Eleonora, Sobur Ali, Sarah E. Schmedes, Brittany Rife Magalis, Simone Marini, Marco Salemi, Jason Blanton, and Taj Azarian. 2022. "Early Emergence Phase of SARS-CoV-2 Delta Variant in Florida, US" Viruses 14, no. 4: 766. https://doi.org/10.3390/v14040766
APA StyleCella, E., Ali, S., Schmedes, S. E., Rife Magalis, B., Marini, S., Salemi, M., Blanton, J., & Azarian, T. (2022). Early Emergence Phase of SARS-CoV-2 Delta Variant in Florida, US. Viruses, 14(4), 766. https://doi.org/10.3390/v14040766