
Understanding the Driving Forces That Trigger Mutations in SARS-CoV-2: Mutational Energetics and the Role of Arginine Blockers in COVID-19 Therapy

 , ,
, ,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Prediction of Changes in Binding Affinity of the RBD-ACE2 Complex
2.2. Molecular Simulations
2.3. Preparation of Ligands
2.4. Molecular Docking
2.5. Construction of a Full-Sequence S-Protein Model for Calculation of Proteo-Stabilities
2.6. Sartan Structures and Pharmacophores
3. Results
3.1. Relationship of Virion Binding to Residue Hydropathies
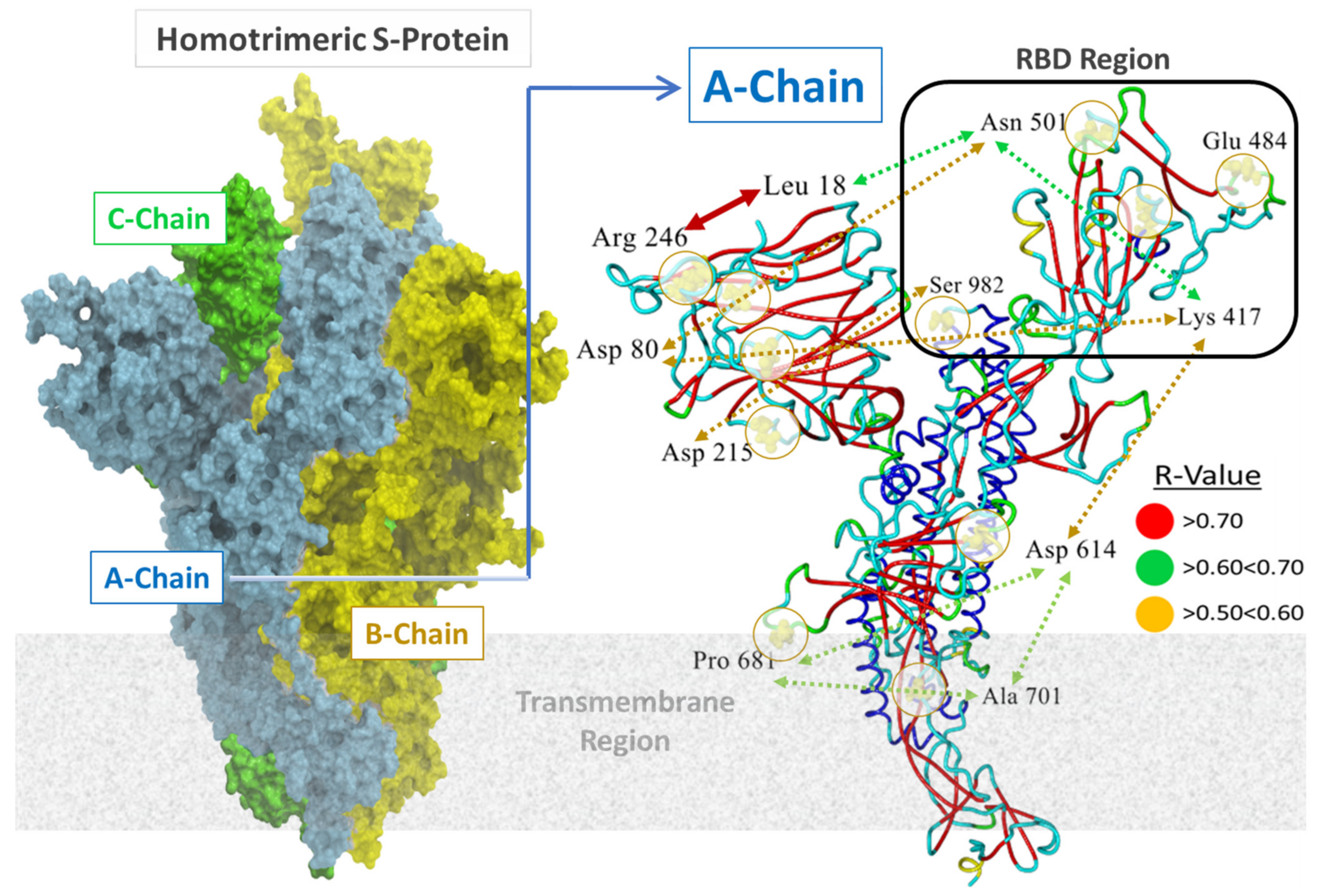
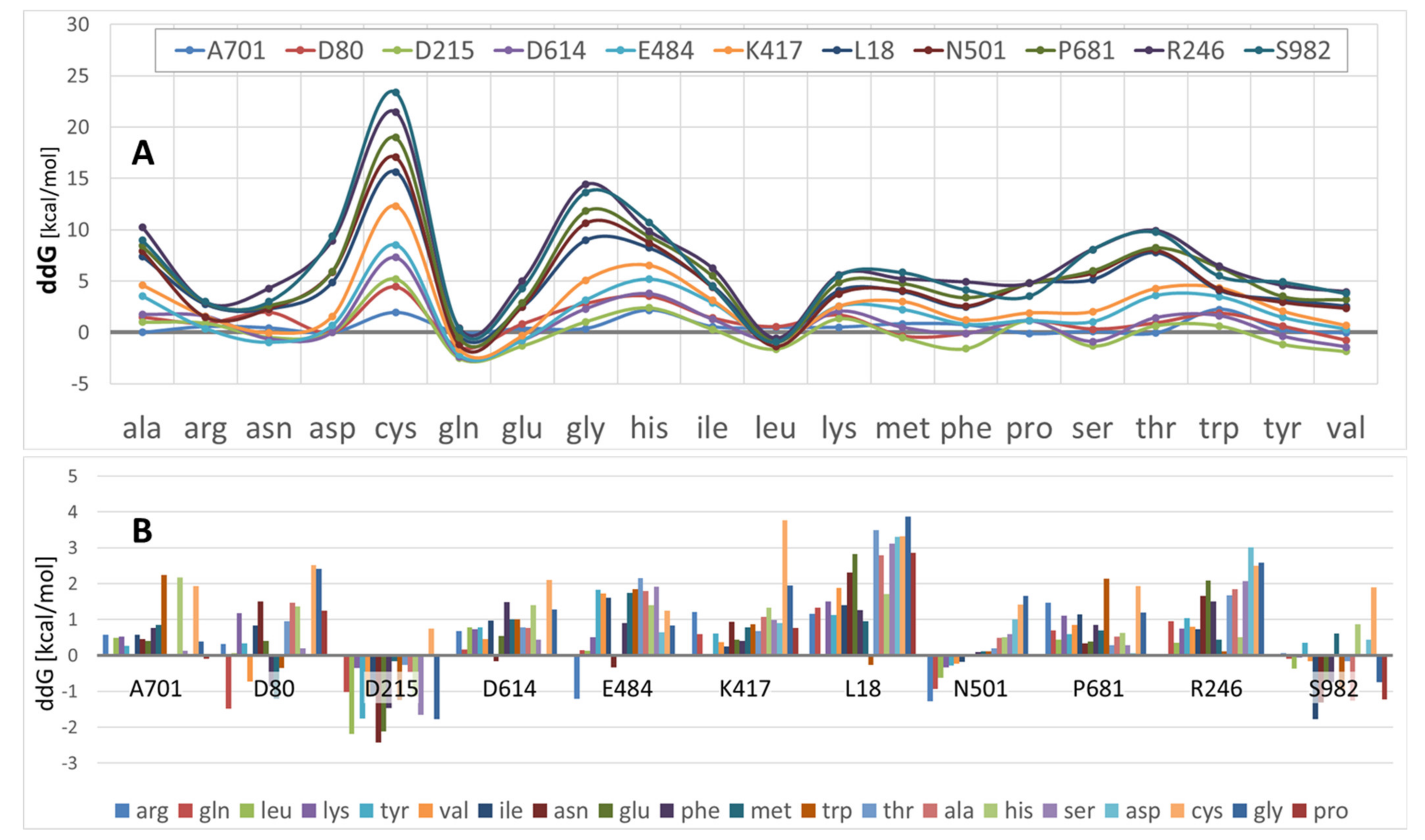
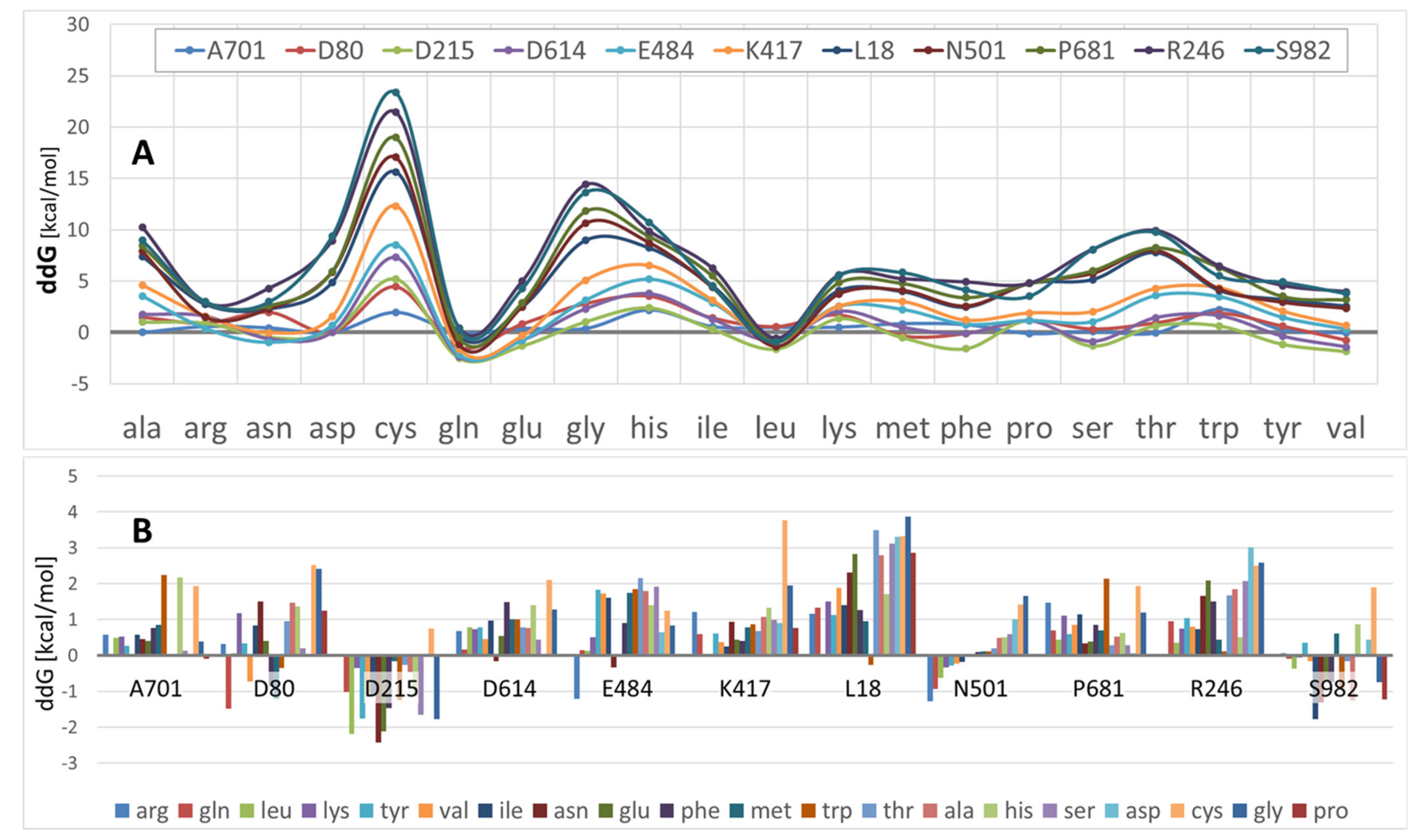
3.2. S-Protein Free-Energy Stability Perturbations Computed for Selected SARS-CoV-2 Mutants
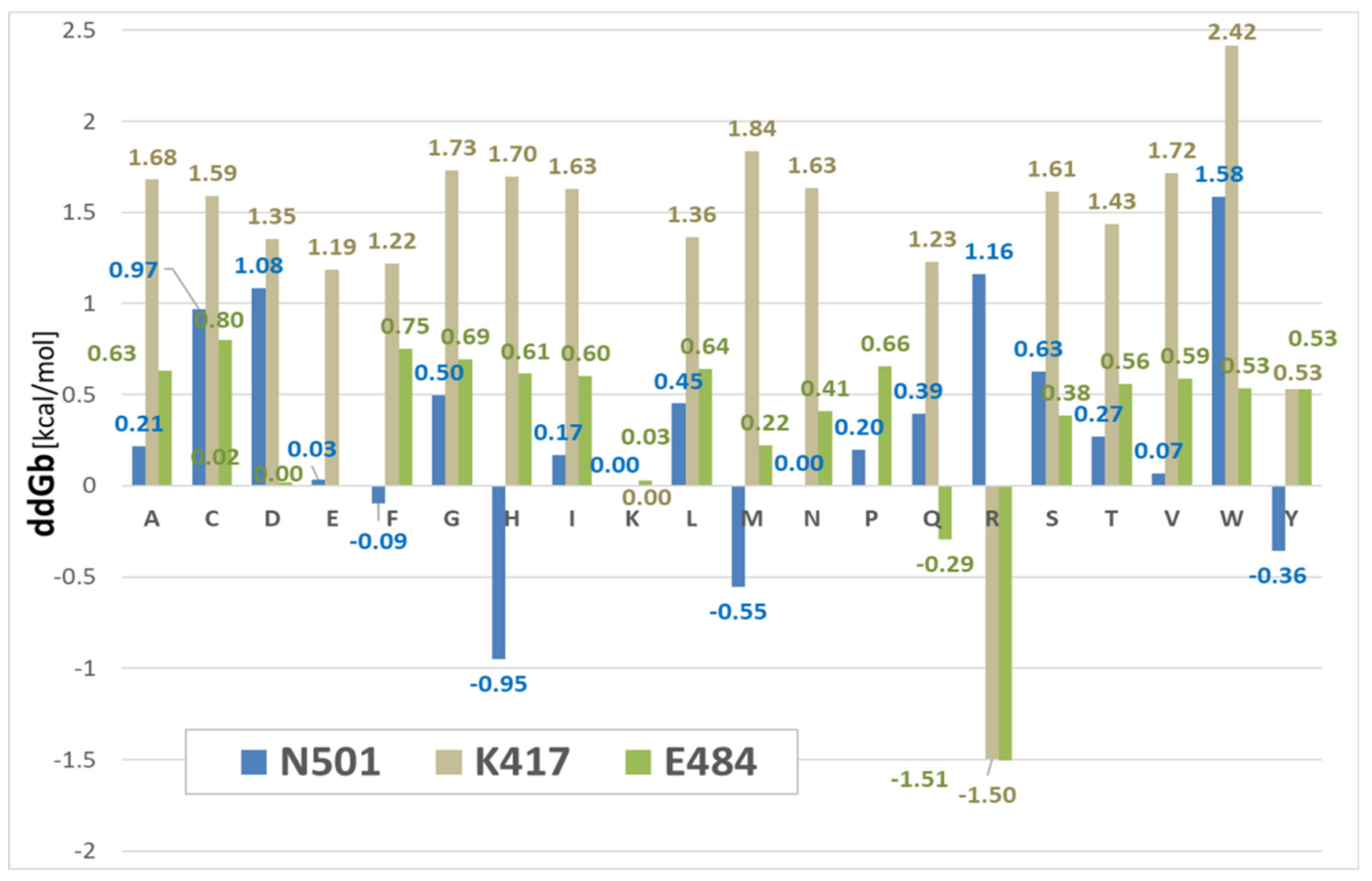
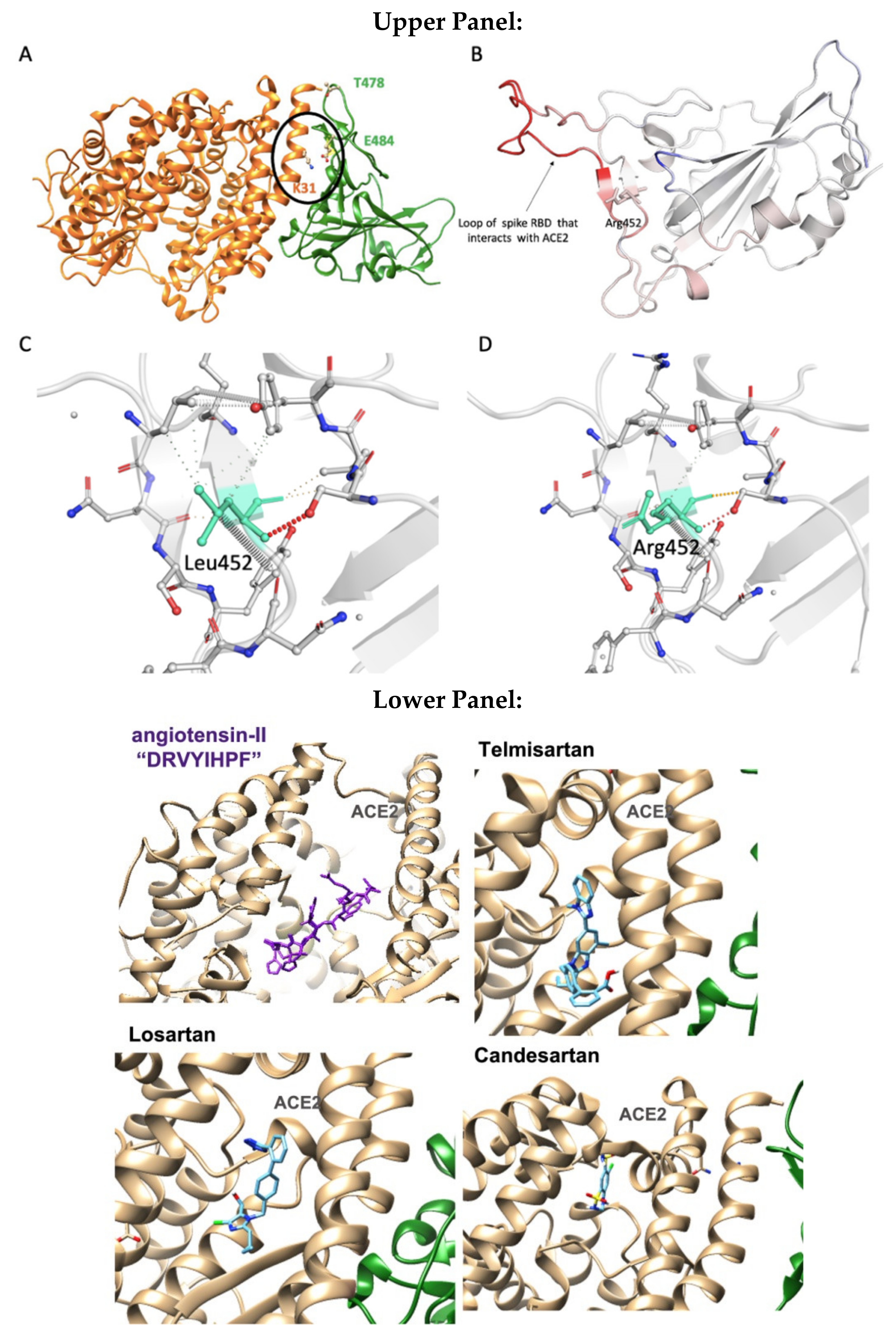
3.3. Binding Free-Energy Calculations between SARS-CoV-2 RBD and Human ACE2 Receptor
3.4. MD Simulations: Binding Energies between SARS-CoV-2 RBD and Human ACE2 Receptor
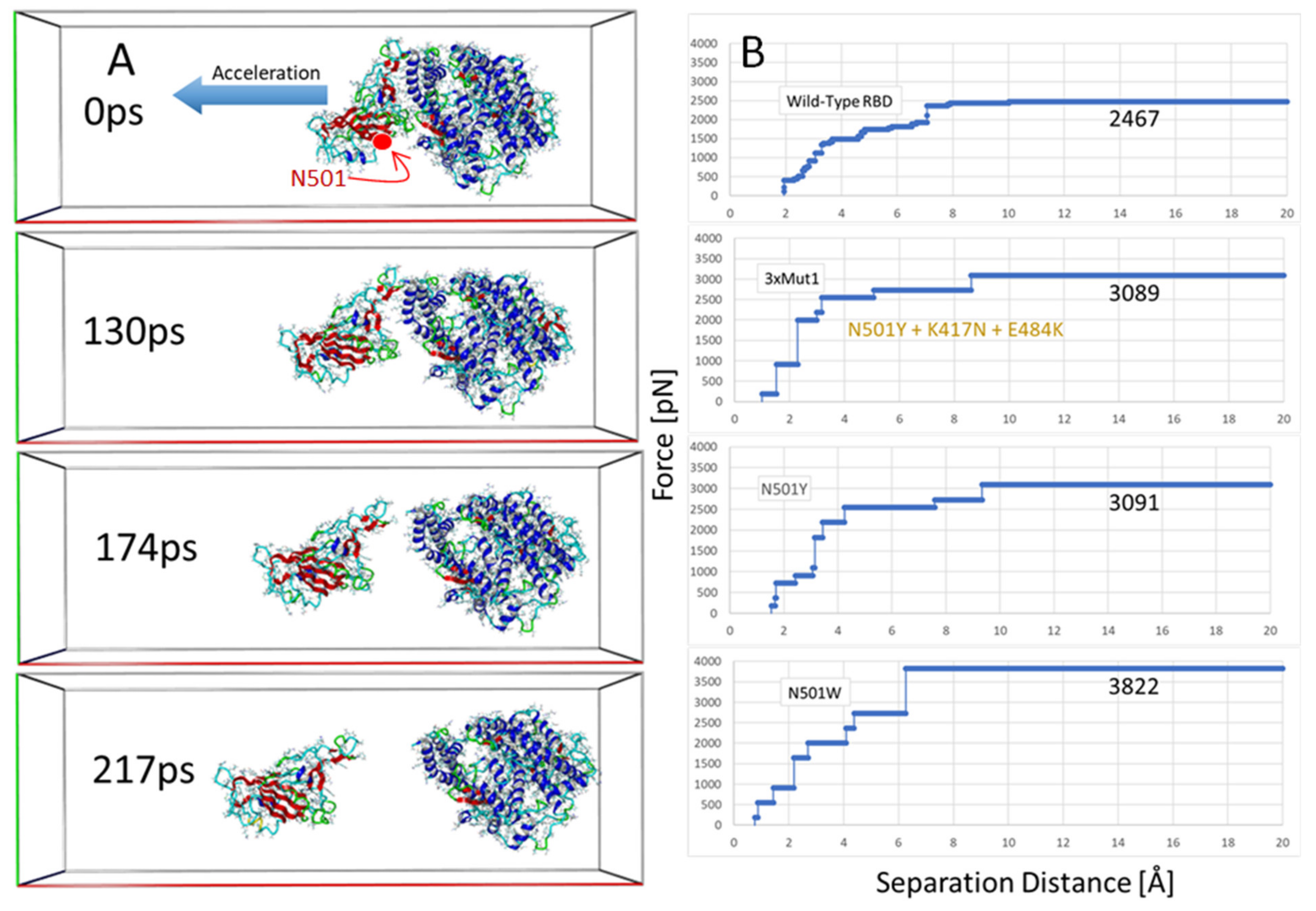
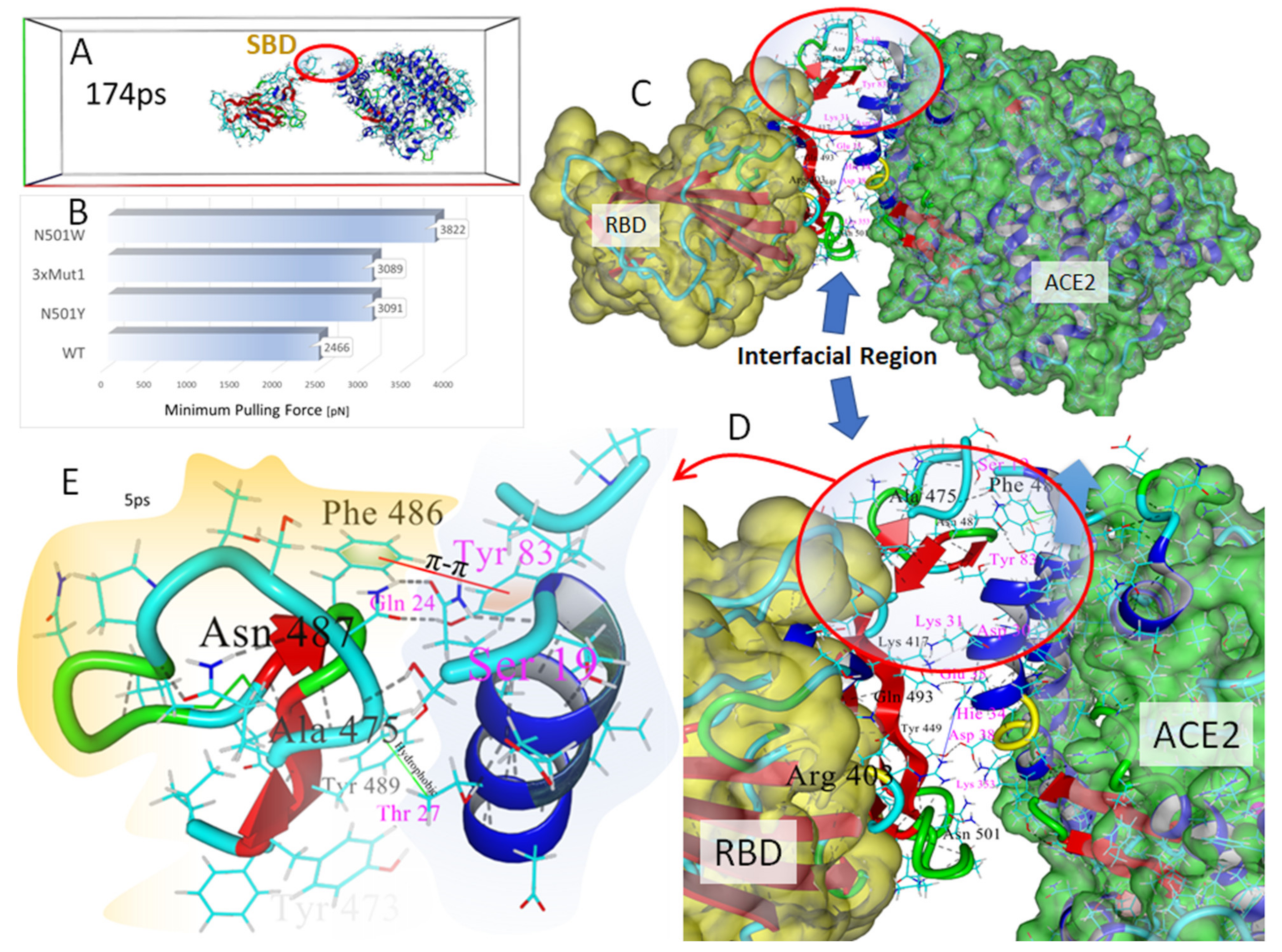
3.5. Steered Molecular Dynamics: RBD-ACE2 Dissociation Behavior
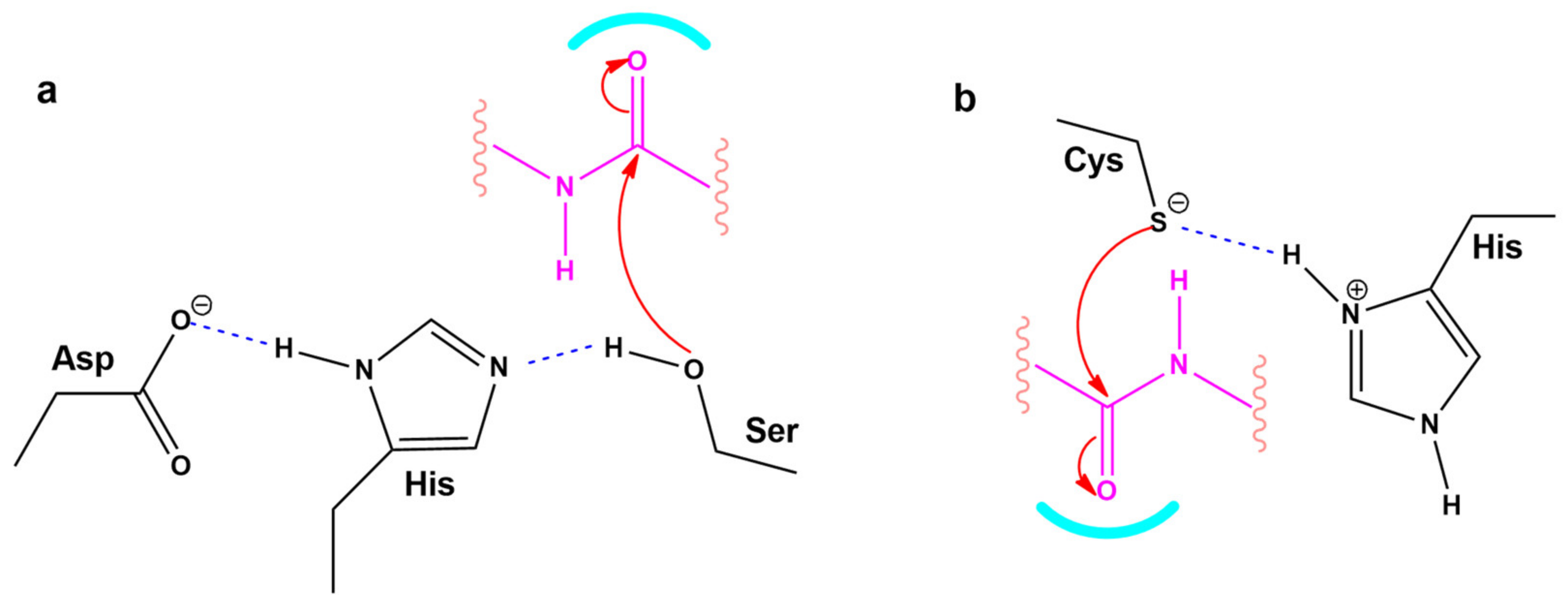
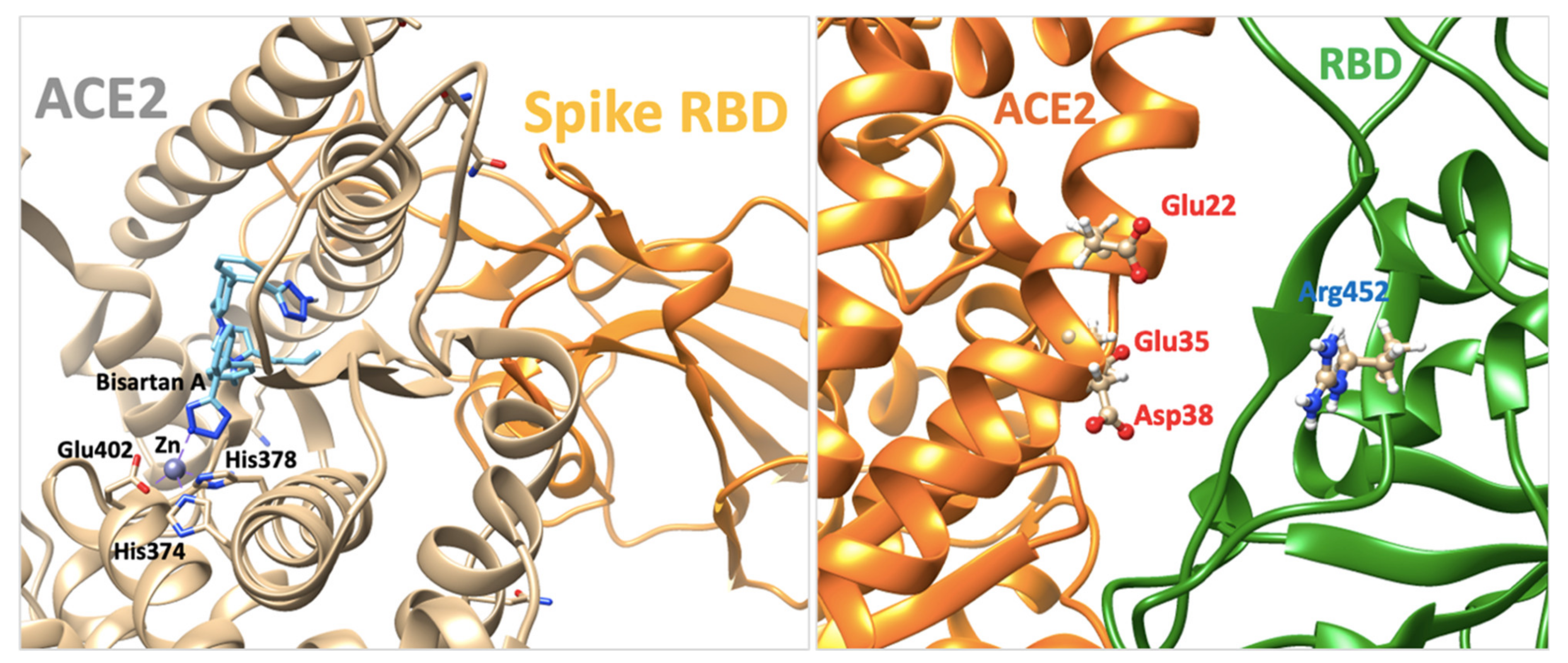
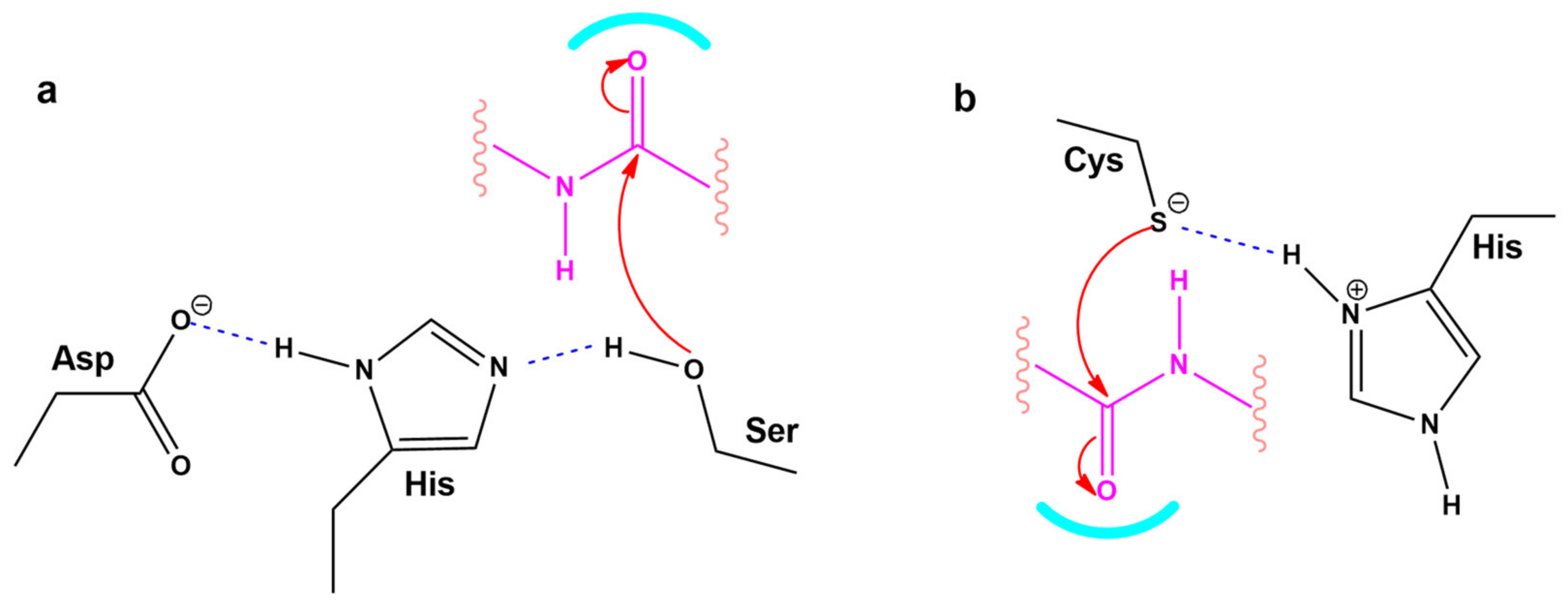
3.6. Modeling the Role of Arginine and the Charge Relay System
3.7. Normal-Mode Molecular Dynamics
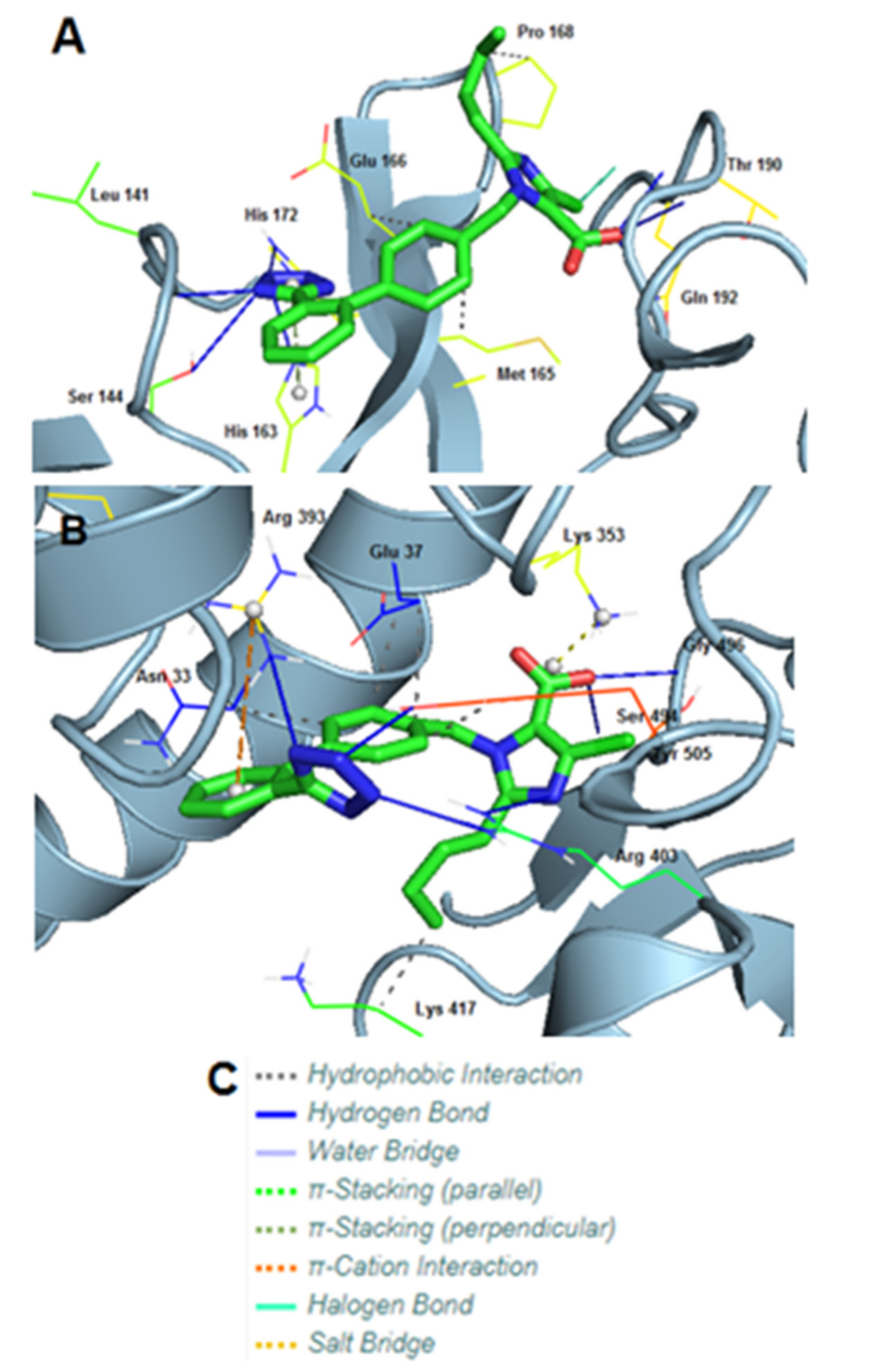
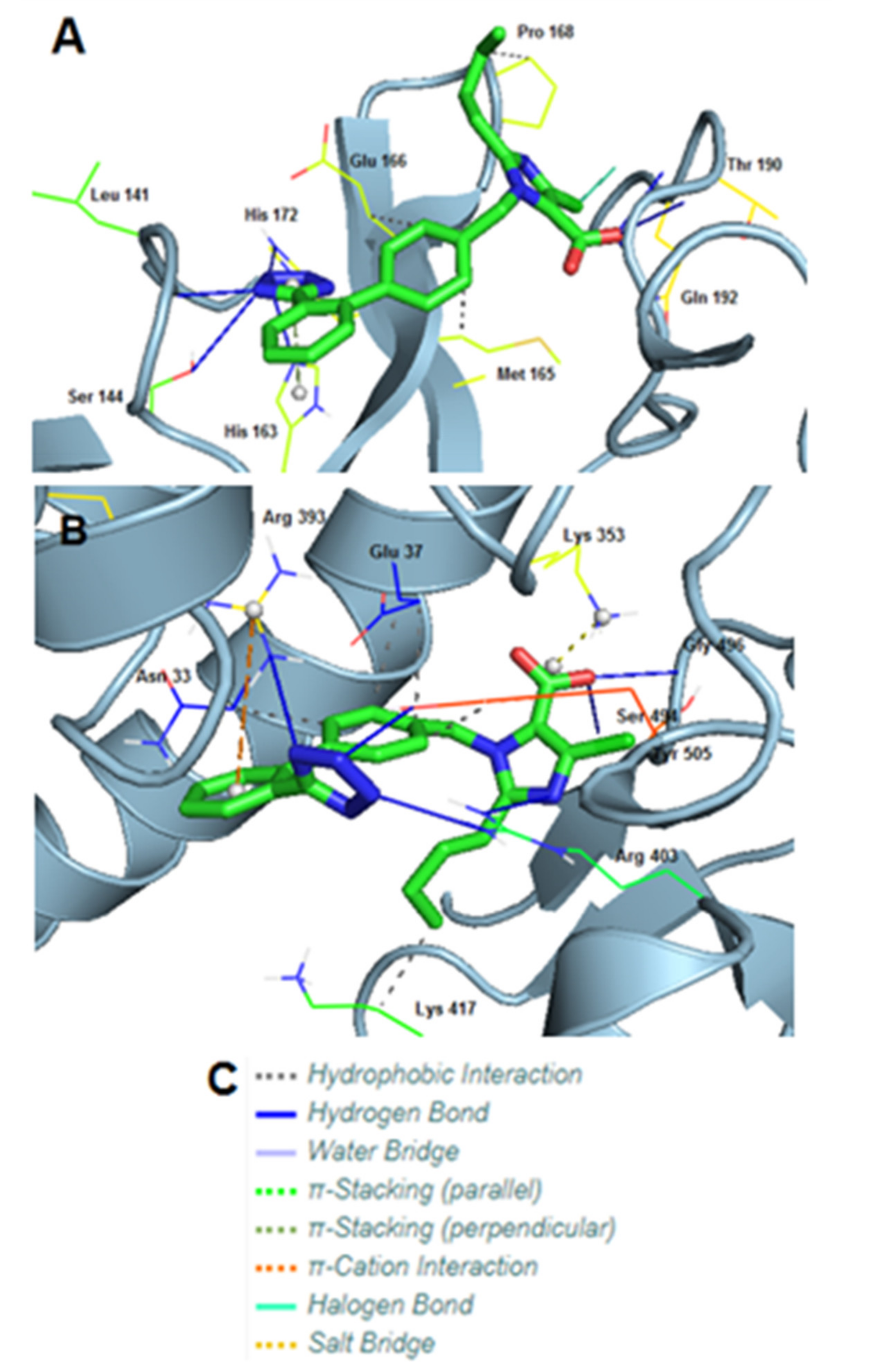
3.8. Molecular Interactions of EXP3174 with Mpro and Spike Protein
4. Discussion
4.1. Dominating Mutations
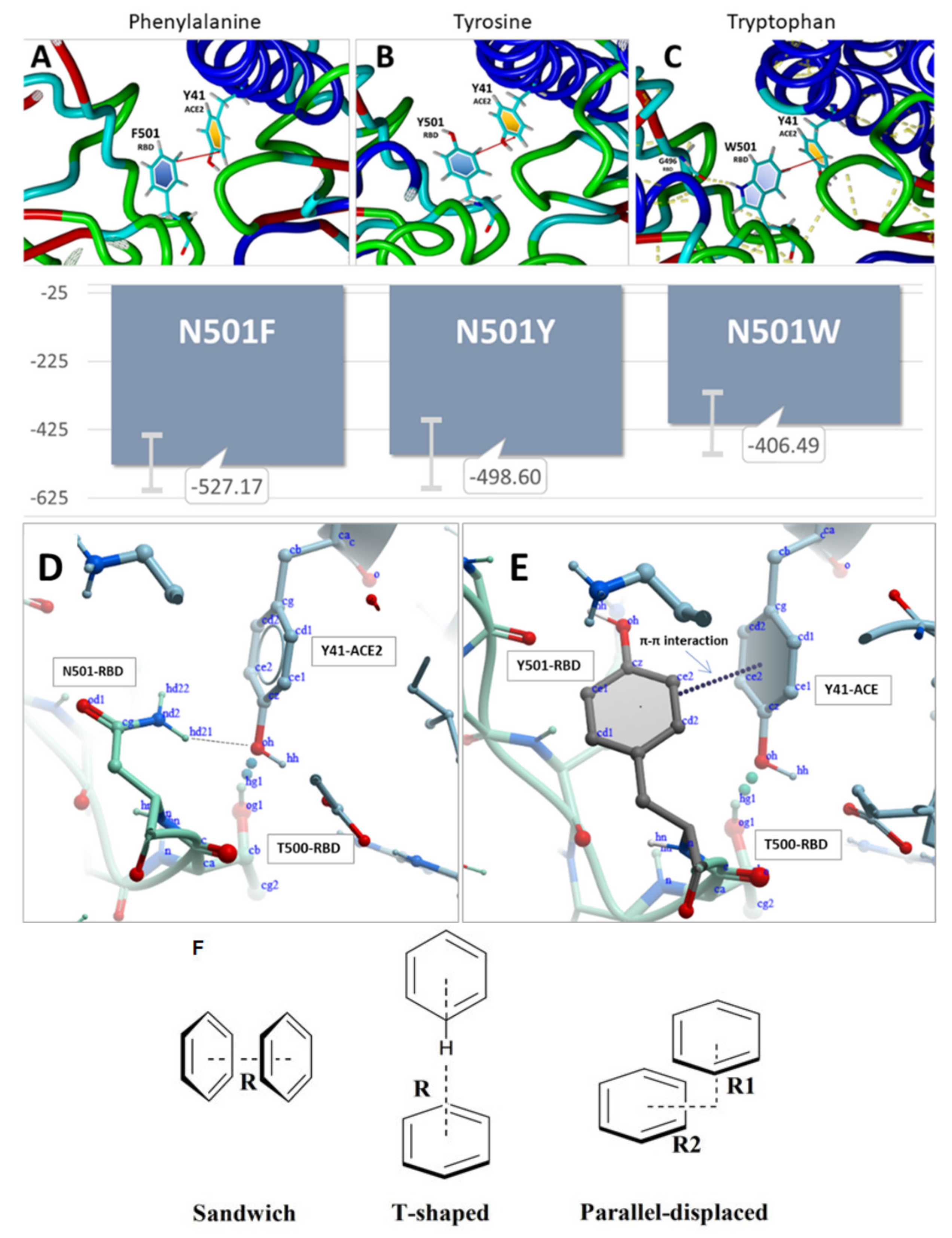
4.2. Mutations and π-π Interactions
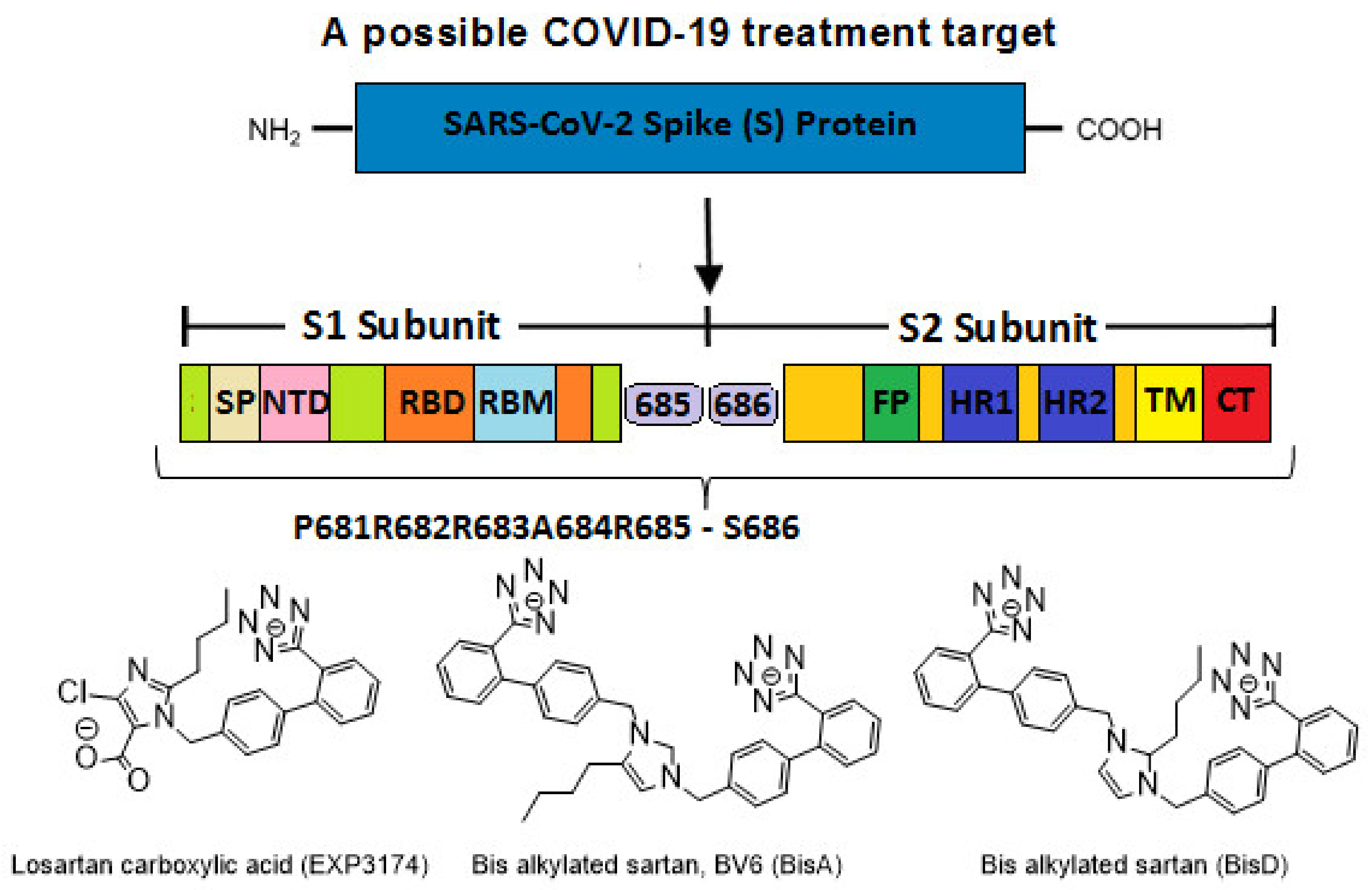
4.3. Targets for SARS-CoV-2 Treatment
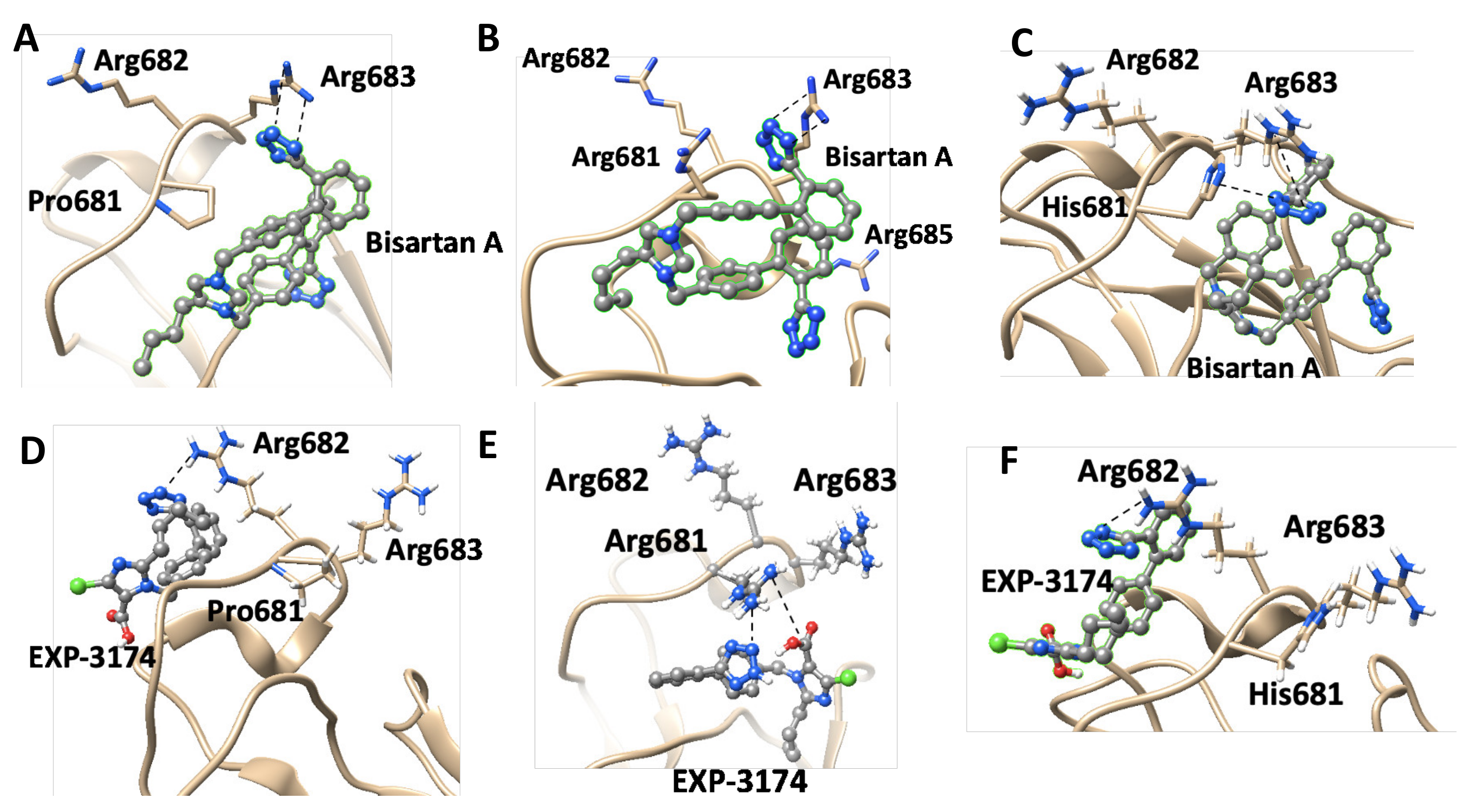
4.4. P681R Delta vs. P681 Version
4.5. CRS Mechanisms in AngII and SARS-CoV-2 Proteases
4.6. ARBs Could Bind to the Basic Arginine Rich Cavity Loop of SARS-CoV-2
4.7. ARBs as Potential RBD/ACE2 Blockers
4.8. Bisartans and the Role of Tetrazole
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.-Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e899. [Google Scholar] [CrossRef] [PubMed]
- Nikhra, V. Evolving Patterns in COVID-19: The Virus, its Variants and Infectivity-cum-Virulence. Biomed. J. Sci. Tech. Res. 2021, 33. [Google Scholar] [CrossRef]
- Sinha, S.; Wang, S.M. Classification of VUS and unclassified variants in BRCA1 BRCT repeats by molecular dynamics simulation. Comput. Struct. Biotechnol. J. 2020, 18, 723–736. [Google Scholar] [CrossRef]
- Ali, A.; Vijayan, R. Dynamics of the ACE2–SARS-CoV-2/SARS-CoV spike protein interface reveal unique mechanisms. Sci. Rep. 2020, 10, 14214. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Simonetti, F.L.; Goncearenco, A.; Panchenko, A.R. MutaBind estimates and interprets the effects of sequence variants on protein–protein interactions. Nucl. Acids Res. 2016, 44, W494–W501. [Google Scholar] [CrossRef] [Green Version]
- Oulas, A.; Richter, J.; Zanti, M.; Tomazou, M.; Michailidou, K.; Christodoulou, K.; Christodoulou, C.; Spyrou, G.M. In depth analysis of Cyprus-specific mutations of SARS-CoV-2 strains using computational approaches. BMC Genom. Data 2021, 22, 48. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera?A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein–ligand interaction profiler. Nucl. Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef] [PubMed]
- Pommié, C.; Levadoux, S.; Sabatier, R.; Lefranc, G.; Lefranc, M.-P. IMGT standardized criteria for statistical analysis of immunoglobulin V-REGION amino acid properties. J. Mol. Recogn. 2004, 17, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, H.; Moore, G.J.; Mavromoustakos, T.; Tsiodras, S.; Ligielli, I.; Kelaidonis, K.; Chasapis, C.T.; Kate Gadanec, L.; Zulli, A.; Apostolopoulos, V.; et al. Discovery of a new generation of angiotensin receptor blocking drugs: Receptor mechanisms and in silico binding to enzymes relevant to COVID-19. Comput. Struct. Biotechnol. J. 2022, 20, 2091–2111. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Prot. Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Allouche, A.-R. Gabedit-A graphical user interface for computational chemistry softwares. J. Comput. Chem. 2011, 32, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Bommu, U.D.; Konidala, K.K.; Pamanji, R.; Yeguvapalli, S. Structural Probing, Screening and Structure-Based Drug Repositioning Insights into the Identification of Potential Cox-2 Inhibitors from Selective Coxibs. Interdiscp. Sci. Computat. Life Sci. 2017, 11, 153–169. [Google Scholar] [CrossRef] [PubMed]
- Hetényi, C.; van der Spoel, D. Efficient docking of peptides to proteins without prior knowledge of the binding site. Prot. Sci. 2009, 11, 1729–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keretsu, S.; Bhujbal, S.P.; Cho, S.J. Rational approach toward COVID-19 main protease inhibitors via molecular docking, molecular dynamics simulation and free energy calculation. Sci. Rep. 2020, 10, 17716. [Google Scholar] [CrossRef]
- Nimgampalle, M.; Devanathan, V.; Saxena, A. Screening of Chloroquine, Hydroxychloroquine and its derivatives for their binding affinity to multiple SARS-CoV-2 protein drug targets. J. Biomol. Struct. Dynam. 2020, 39, 4949–4961. [Google Scholar] [CrossRef]
- Patel, A.; Rajendran, M.; Shah, A.; Patel, H.; Pakala, S.B.; Karyala, P. Virtual screening of curcumin and its analogs against the spike surface glycoprotein of SARS-CoV-2 and SARS-CoV. J. Biomol. Struct. Dynam. 2021, 1–9. [Google Scholar] [CrossRef]
- Durdagi, S.; Avsar, T.; Orhan, M.D.; Serhatli, M.; Balcioglu, B.K.; Ozturk, H.U.; Kayabolen, A.; Cetin, Y.; Aydinlik, S.; Bagci-Onder, T.; et al. The neutralization effect of montelukast on SARS-CoV-2 is shown by multiscale in silico simulations and combined in vitro studies. Mol. Ther. 2022, 30, 963–974. [Google Scholar] [CrossRef]
- Isaac-Lam, M.F. Molecular modeling of the interaction of ligands with ACE2–SARS-CoV-2 spike protein complex. In Silico Pharmacol. 2021, 9, 55. [Google Scholar] [CrossRef]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]
- Laffeber, C.; de Koning, K.; Kanaar, R.; Lebbink, J.H.G. Experimental Evidence for Enhanced Receptor Binding by Rapidly Spreading SARS-CoV-2 Variants. J. Mol. Biol. 2021, 433, 167058. [Google Scholar] [CrossRef]
- Tong, B.; Tian, F.; Sun, L.; Shi, S.; Zheng, B.; Wang, Z.; Dong, X.; Zheng, P. N501Y mutation of spike protein in SARS-CoV-2 strengthens its binding to receptor ACE2. eLife 2021, 10, e69091. [Google Scholar] [CrossRef]
- Liu, L.; Iketani, S.; Guo, Y.; Chan, J.F.W.; Wang, M.; Liu, L.; Luo, Y.; Chu, H.; Huang, Y.; Nair, M.S.; et al. Striking antibody evasion manifested by the Omicron variant of SARS-CoV-2. Nature 2021, 602, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Tam, B.; Wang, S.M. RBD Double Mutations of SARS-CoV-2 Strains Increase Transmissibility through Enhanced Interaction between RBD and ACE2 Receptor. Viruses 2021, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Fratev, F. N501Y and K417N Mutations in the Spike Protein of SARS-CoV-2 Alter the Interactions with Both hACE2 and Human-Derived Antibody: A Free Energy of Perturbation Retrospective Study. J. Chem. Inform. Model. 2021, 61, 6079–6084. [Google Scholar] [CrossRef] [PubMed]
- Luan, B.; Wang, H.; Huynh, T. Enhanced binding of the N501Y-mutated SARS-CoV-2 spike protein to the human ACE2 receptor: Insights from molecular dynamics simulations. FEBS Lett. 2021, 595, 1454–1461. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, M.; Gao, J. Enhanced receptor binding of SARS-CoV-2 through networks of hydrogen-bonding and hydrophobic interactions. Proc. Natl. Acad. Sci. USA 2020, 117, 13967–13974. [Google Scholar] [CrossRef]
- Socher, E.; Heger, L.; Paulsen, F.; Zunke, F.; Arnold, P. Molecular dynamics simulations of the delta and omicron SARS-CoV-2 spike–ACE2 complexes reveal distinct changes between both variants. Comput. Struct. Biotechnol. J. 2022, 20, 1168–1176. [Google Scholar] [CrossRef]
- Kim, T.; Rhee, A.; Yip, C.M. Force-Induced Insulin Dimer Dissociation: A Molecular Dynamics Study. J. Am. Chem. Soc. 2006, 128, 5330–5331. [Google Scholar] [CrossRef]
- Gao, Q.; Lu, C.; Wang, X.-W.; Zhang, J.-W.; Song, Y.; Xue, Y.-L. Molecular dynamics simulation and steered molecular dynamics simulation on irisin dimers. J. Mol. Model. 2018, 24, 95. [Google Scholar] [CrossRef]
- Liu, Z.; Moreira, R.A.; Dujmović, A.; Liu, H.; Yang, B.; Poma, A.B.; Nash, M.A. Mapping Mechanostable Pulling Geometries of a Therapeutic Anticalin/CTLA-4 Protein Complex. Nano Lett. 2021, 22, 179–187. [Google Scholar] [CrossRef]
- Garcia-Manyes, S.; Badilla, C.L.; Alegre-Cebollada, J.; Javadi, Y.; Fernández, J.M. Spontaneous Dimerization of Titin Protein Z1Z2 Domains Induces Strong Nanomechanical Anchoring. J. Biol. Chem. 2012, 287, 20240–20247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Huang, Z.; Lin, L.; Lv, J. Coronavirus Disease 2019 (COVID-19) and Cardiovascular Disease: A Viewpoint on the Potential Influence of Angiotensin-Converting Enzyme Inhibitors/Angiotensin Receptor Blockers on Onset and Severity of Severe Acute Respiratory Syndrome Coronavirus 2 Infection. J. Am. Heart Assoc. 2020, 9, e016219. [Google Scholar] [CrossRef] [PubMed]
- Verdecchia, P.; Cavallini, C.; Spanevello, A.; Angeli, F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 2020, 76, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Qaradakhi, T.; Gadanec, L.K.; McSweeney, K.R.; Tacey, A.; Apostolopoulos, V.; Levinger, I.; Rimarova, K.; Egom, E.E.; Rodrigo, L.; Kruzliak, P.; et al. The potential actions of angiotensin-converting enzyme II (ACE2) activator diminazene aceturate (DIZE) in various diseases. Clin. Exp. Pharmacol. Physiol. 2020, 47, 751–758. [Google Scholar] [CrossRef]
- Zhang, H.; Unal, H.; Desnoyer, R.; Han, G.W.; Patel, N.; Katritch, V.; Karnik, S.S.; Cherezov, V.; Stevens, R.C. Structural Basis for Ligand Recognition and Functional Selectivity at Angiotensin Receptor. J. Biol. Chem. 2015, 290, 29127–29139. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Unal, H.; Gati, C.; Han, G.W.; Liu, W.; Zatsepin, N.A.; James, D.; Wang, D.; Nelson, G.; Weierstall, U.; et al. Structure of the Angiotensin Receptor Revealed by Serial Femtosecond Crystallography. Cell 2015, 161, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Agelis, G.; Resvani, A.; Koukoulitsa, C.; Tůmová, T.; Slaninová, J.; Kalavrizioti, D.; Spyridaki, K.; Afantitis, A.; Melagraki, G.; Siafaka, A.; et al. Rational design, efficient syntheses and biological evaluation of N, N ′-symmetrically bis-substituted butylimidazole analogs as a new class of potent Angiotensin II receptor blockers. Eur. J. Med. Chem. 2013, 62, 352–370. [Google Scholar] [CrossRef] [Green Version]
- Agelis, G.; Resvani, A.; Ntountaniotis, D.; Chatzigeorgiou, P.; Koukoulitsa, C.; Androutsou, M.E.; Plotas, P.; Matsoukas, J.; Mavromoustakos, T.; Čendak, T.; et al. Interactions of the potent synthetic AT1 antagonist analog BV6 with membrane bilayers and mesoporous silicate matrices. Biochim. Biophys. Acta-Biomembr. 2013, 1828, 1846–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaimes, J.A.; André, N.M.; Chappie, J.S.; Millet, J.K.; Whittaker, G.R. Phylogenetic Analysis and Structural Modeling of SARS-CoV-2 Spike Protein Reveals an Evolutionary Distinct and Proteolytically Sensitive Activation Loop. J. Mol. Biol. 2020, 432, 3309–3325. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784.e775. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neitzel, J.J. Enzyme Catalysis: The Serine Proteases. Nat. Educ. 2010, 3, 21. [Google Scholar]
- McGaughey, G.B.; Gagné, M.; Rappé, A.K. π-Stacking Interactions. J. Biol. Chem. 1998, 273, 15458–15463. [Google Scholar] [CrossRef] [Green Version]
- Martinez, C.R.; Iverson, B.L. Rethinking the term “pi-stacking”. Chem. Sci. 2012, 3, 2191. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Wang, R.; Wang, M.; Wei, G.W. Mutations Strengthened SARS-CoV-2 Infectivity. J. Mol. Biol. 2020, 432, 5212–5226. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Johnson, B.A.; Xia, H.; Ku, Z.; Schindewolf, C.; Widen, S.G.; An, Z.; Weaver, S.C.; Menachery, V.D.; et al. Delta spike P681R mutation enhances SARS-CoV-2 fitness over Alpha variant. bioRxiv 2021. preprint. [Google Scholar] [CrossRef]
- Blow, D.M.; Birktoft, J.J.; Hartley, B.S. Role of a Buried Acid Group in the Mechanism of Action of Chymotrypsin. Nature 1969, 221, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Moore, G.J.; Matsoukas, J.M. Angiotensin as a model for hormone–receptor interactions. Biosci. Rep. 1985, 5, 407–416. [Google Scholar] [CrossRef]
- Moreno, M.; Bataller, R. Cytokines and Renin-Angiotensin System Signaling in Hepatic Fibrosis. Clin. Liver Dis. 2008, 12, 825–852. [Google Scholar] [CrossRef]
- Ruiz-Ortega, M.; Ruperez, M.; Lorenzo, O.; Esteban, V.; Blanco, J.; Mezzano, S.; Egido, J. Angiotensin II regulates the synthesis of proinflammatory cytokines and chemokines in the kidney. Kidney Int. 2002, 62, S12–S22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, J.C.; Fadl, S.; Villanueva, A.J.; Rabeh, W.M. Catalytic Dyad Residues His41 and Cys145 Impact the Catalytic Activity and Overall Conformational Fold of the Main SARS-CoV-2 Protease 3-Chymotrypsin-Like Protease. Front. Chem. 2021, 9, 692168. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 M pro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 6575. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, B.; Batool, M.; Ain, Q.u.; Kim, M.S.; Choi, S. Exploring the Binding Mechanism of PF-07321332 SARS-CoV-2 Protease Inhibitor through Molecular Dynamics and Binding Free Energy Simulations. Int. J. Mol. Sci. 2021, 22, 9124. [Google Scholar] [CrossRef]
- Berteotti, A.; Vacondio, F.; Lodola, A.; Bassi, M.; Silva, C.; Mor, M.; Cavalli, A. Predicting the Reactivity of Nitrile-Carrying Compounds with Cysteine: A Combined Computational and Experimental Study. ACS Med. Chem. Lett. 2014, 5, 501–505. [Google Scholar] [CrossRef]
- Lubinski, B.; Fernandes, M.H.V.; Frazier, L.; Tang, T.; Daniel, S.; Diel, D.G.; Jaimes, J.A.; Whittaker, G.R. Functional evaluation of the P681H mutation on the proteolytic activation of the SARS-CoV-2 variant B.1.1.7 (Alpha) spike. iScience 2022, 25, 103589. [Google Scholar] [CrossRef]
- Matsoukas, J.M.; Agelis, G.; Hondrelis, J.; Yamdagni, R.; Wu, Q.; Ganter, R.; Moore, D.; Moore, G.J.; Smith, J.R. Synthesis and biological activities of angiotensin II, sarilesin, and sarmesin analogs containing Aze or Pip at position 7. J. Med. Chem. 2002, 36, 904–911. [Google Scholar] [CrossRef]
- Hondrelis, J.; Lonergan, G.; Voliotis, S.; Matsoukas, J. One pot synthesis and conformation of N-t-butyloxycarbonyl, O-Phenacyl derivatives of proline and other secondary amino acids. Tetrahedron 1990, 46, 565–576. [Google Scholar] [CrossRef]
- Moore, G.J.; Smitht, J.R.; Baylis, B.W.; Matsoukas, J.M. Design and Pharmacology of Peptide Mimetics. Adv. Pharmacol. 1995, 33, 91–141. [Google Scholar] [CrossRef]
- Lubbe, L.; Cozier, G.E.; Oosthuizen, D.; Acharya, K.R.; Sturrock, E.D. ACE2 and ACE: Structure-based insights into mechanism, regulation and receptor recognition by SARS-CoV. Clin. Sci. 2020, 134, 2851–2871. [Google Scholar] [CrossRef]
- Takezako, T.; Unal, H.; Karnik, S.S.; Node, K. Current topics in angiotensin II type 1 receptor research: Focus on inverse agonism, receptor dimerization and biased agonism. Pharmacol. Res. 2017, 123, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef] [PubMed]
- Duarte, M.; Pelorosso, F.; Nicolosi, L.N.; Victoria Salgado, M.; Vetulli, H.; Aquieri, A.; Azzato, F.; Castro, M.; Coyle, J.; Davolos, I.; et al. Telmisartan for treatment of Covid-19 patients: An open multicenter randomized clinical trial. EClinicalMedicine 2021, 37, 100962. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Xiao, G.; Zhang, J.; He, X.; Ou, M.; Bi, J.; Yang, R.; Di, W.; Wang, Z.; Li, Z.; et al. Renin-angiotensin system inhibitors improve the clinical outcomes of COVID-19 patients with hypertension. Emerg. Microb. Infect. 2020, 9, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Vaduganathan, M.; Vardeny, O.; Michel, T.; McMurray, J.J.V.; Pfeffer, M.A.; Solomon, S.D. Renin–Angiotensin–Aldosterone System Inhibitors in Patients with Covid-19. N. Engl. J. Med. 2020, 382, 1653–1659. [Google Scholar] [CrossRef]
- Zhang, P.; Zhu, L.; Cai, J.; Lei, F.; Qin, J.-J.; Xie, J.; Liu, Y.-M.; Zhao, Y.-C.; Huang, X.; Lin, L.; et al. Association of Inpatient Use of Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers with Mortality Among Patients with Hypertension Hospitalized with COVID-19. Circ. Res. 2020, 126, 1671–1681. [Google Scholar] [CrossRef]
- Moore, G.J.; Pires, J.M.; Kelaidonis, K.; Gadanec, L.K.; Zulli, A.; Apostolopoulos, V.; Matsoukas, J.M. Receptor Interactions of Angiotensin II and Angiotensin Receptor Blockers—Relevance to COVID-19. Biomolecules 2021, 11, 979. [Google Scholar] [CrossRef]
- Matsoukas, J.M.; Hondrelis, J.; Keramida, M.; Mavromoustakos, T.; Makriyannis, A.; Yamdagni, R.; Wu, Q.; Moore, G.J. Role of the NH2-terminal domain of angiotensin II (ANG II) and [Sar1]angiotensin II on conformation and activity. NMR evidence for aromatic ring clustering and peptide backbone folding compared with [des-1,2,3]angiotensin II. J. Biol. Chem. 1994, 269, 5303–5312. [Google Scholar] [CrossRef]
- Yamamoto, K.; Tanabe, N.; Takahashi, Y.; Naito, A.; Sekine, A.; Suda, R.; Jujo Sanada, T.; Sugiura, T.; Shigeta, A.; Sakao, S.; et al. Characteristics of patients meeting the new definition of pre-capillary pulmonary hypertension (Nice 2018) in a single Japanese pulmonary hypertension center. BMC Pulm. Med. 2021, 21, 260. [Google Scholar] [CrossRef]
- Jhund, P.S.; McMurray, J.J.V. The neprilysin pathway in heart failure: A review and guide on the use of sacubitril/valsartan. Heart 2016, 102, 1342–1347. [Google Scholar] [CrossRef] [Green Version]
- Schiering, N.; D’Arcy, A.; Villard, F.; Ramage, P.; Logel, C.; Cumin, F.; Ksander, G.M.; Wiesmann, C.; Karki, R.G.; Mogi, M. Structure of neprilysin in complex with the active metabolite of sacubitril. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Örd, M.; Faustova, I.; Loog, M. The sequence at Spike S1/S2 site enables cleavage by furin and phospho-regulation in SARS-CoV2 but not in SARS-CoV1 or MERS-CoV. Sci. Rep. 2020, 10, 27909. [Google Scholar] [CrossRef] [PubMed]
- Evangelou, K.; Veroutis, D.; Paschalaki, K.; Foukas, P.G.; Lagopati, N.; Dimitriou, M.; Papaspyropoulos, A.; Konda, B.; Hazapis, O.; Polyzou, A.; et al. Pulmonary infection by SARS-CoV-2 induces senescence accompanied by an inflammatory phenotype in severe COVID-19: Possible implications for viral mutagenesis. Eur. Resp. J. 2022, 59, 2102951. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.K.; Tan, T.J.C.; Narayanan, K.K.; Procko, E. An engineered decoy receptor for SARS-CoV-2 broadly binds protein S sequence variants. Sci. Adv. 2021, 7, 8. [Google Scholar] [CrossRef]
- Glasgow, A.; Glasgow, J.; Limonta, D.; Solomon, P.; Lui, I.; Zhang, Y.; Nix, M.A.; Rettko, N.J.; Zha, S.; Yamin, R.; et al. Engineered ACE2 receptor traps potently neutralize SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 28046–28055. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Best Scoring Pose Bound to ACE2-Spike RBD Complex (kcal/mol) |
|---|---|
| Candesartan | −7.1 |
| Losartan | −8.1 |
| Telmisartan | −9.5 |
| BisA | −9.5 |
| AngII | −12.4 |
| I | V | L | F | C | M | A | W | G | T | S | Y | P | H | N | D | Q | E | K | R |
| 4.5 | 4.2 | 3.8 | 2.8 | 2.5 | 1.9 | 1.8 | −0.9 | −0.4 | −0.7 | −0.8 | −1.3 | −1.6 | −3.2 | −3.5 | −3.5 | −3.5 | −3.5 | −3.5 | −4.5 |
| Hydrophobic | Neutral | Hydrophilic | |||||||||||||||||
| Type of Interactions | Amino Acids of Mpro Involved and Distance of Interactions (A°) | |
|---|---|---|
| Hydrogen Bonds | Leu141 | 1.91 |
| Ser144 | 3.40 | |
| His163 | 2.00 | |
| His172 | 3.61 | |
| Thr190 | 2.09 | |
| Thr190 | 1.86 | |
| Gln192 | 1.98 | |
| Hydrophobic Interactions | Met165 | 3.25 |
| Glu166 | 3.97 | |
| Pro168 | 3.82 | |
| π-Stacking (Stacking Type T) | His163 | 4.11 |
| Halogen Bonds | Gln192 | 3.61 |
| Type of Interactions | Amino Acids of S-RBD Involved and Distance of Interactions (A°) | |
|---|---|---|
| Hydrogen Bonds | Arg393 | 2.47 |
| Arg403 | 2.42 | |
| Arg403 | 2.12 | |
| Ser494 | 3.38 | |
| Gly496 | 1.83 | |
| Tyr505 | 1.94 | |
| Hydrophobic Interactions | Asn33 | 3.94 |
| Glu37 | 3.75 | |
| Glu37 | 3.45 | |
| Lys417 | 3.63 | |
| Tyr505 | 3.50 | |
| π-Cation Interactions | Arg393 | 5.33 |
| Salt Bridges | Lys353 | 2.73 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ridgway, H.; Chasapis, C.T.; Kelaidonis, K.; Ligielli, I.; Moore, G.J.; Gadanec, L.K.; Zulli, A.; Apostolopoulos, V.; Mavromoustakos, T.; Matsoukas, J.M. Understanding the Driving Forces That Trigger Mutations in SARS-CoV-2: Mutational Energetics and the Role of Arginine Blockers in COVID-19 Therapy. Viruses 2022, 14, 1029. https://doi.org/10.3390/v14051029
Ridgway H, Chasapis CT, Kelaidonis K, Ligielli I, Moore GJ, Gadanec LK, Zulli A, Apostolopoulos V, Mavromoustakos T, Matsoukas JM. Understanding the Driving Forces That Trigger Mutations in SARS-CoV-2: Mutational Energetics and the Role of Arginine Blockers in COVID-19 Therapy. Viruses. 2022; 14(5):1029. https://doi.org/10.3390/v14051029
Chicago/Turabian StyleRidgway, Harry, Christos T. Chasapis, Konstantinos Kelaidonis, Irene Ligielli, Graham J. Moore, Laura Kate Gadanec, Anthony Zulli, Vasso Apostolopoulos, Thomas Mavromoustakos, and John M. Matsoukas. 2022. "Understanding the Driving Forces That Trigger Mutations in SARS-CoV-2: Mutational Energetics and the Role of Arginine Blockers in COVID-19 Therapy" Viruses 14, no. 5: 1029. https://doi.org/10.3390/v14051029
APA StyleRidgway, H., Chasapis, C. T., Kelaidonis, K., Ligielli, I., Moore, G. J., Gadanec, L. K., Zulli, A., Apostolopoulos, V., Mavromoustakos, T., & Matsoukas, J. M. (2022). Understanding the Driving Forces That Trigger Mutations in SARS-CoV-2: Mutational Energetics and the Role of Arginine Blockers in COVID-19 Therapy. Viruses, 14(5), 1029. https://doi.org/10.3390/v14051029