
The Contribution of Viral Proteins to the Synergy of Influenza and Bacterial Co-Infection

 , , , and
, , , and
Abstract
:1. Introduction
2. Pathogenesis of Co-Infection by IAV and Streptococcus pneumoniae
2.1. Viral Influenza Infection
2.2. Bacterial Infection with Streptococcus pneumoniae
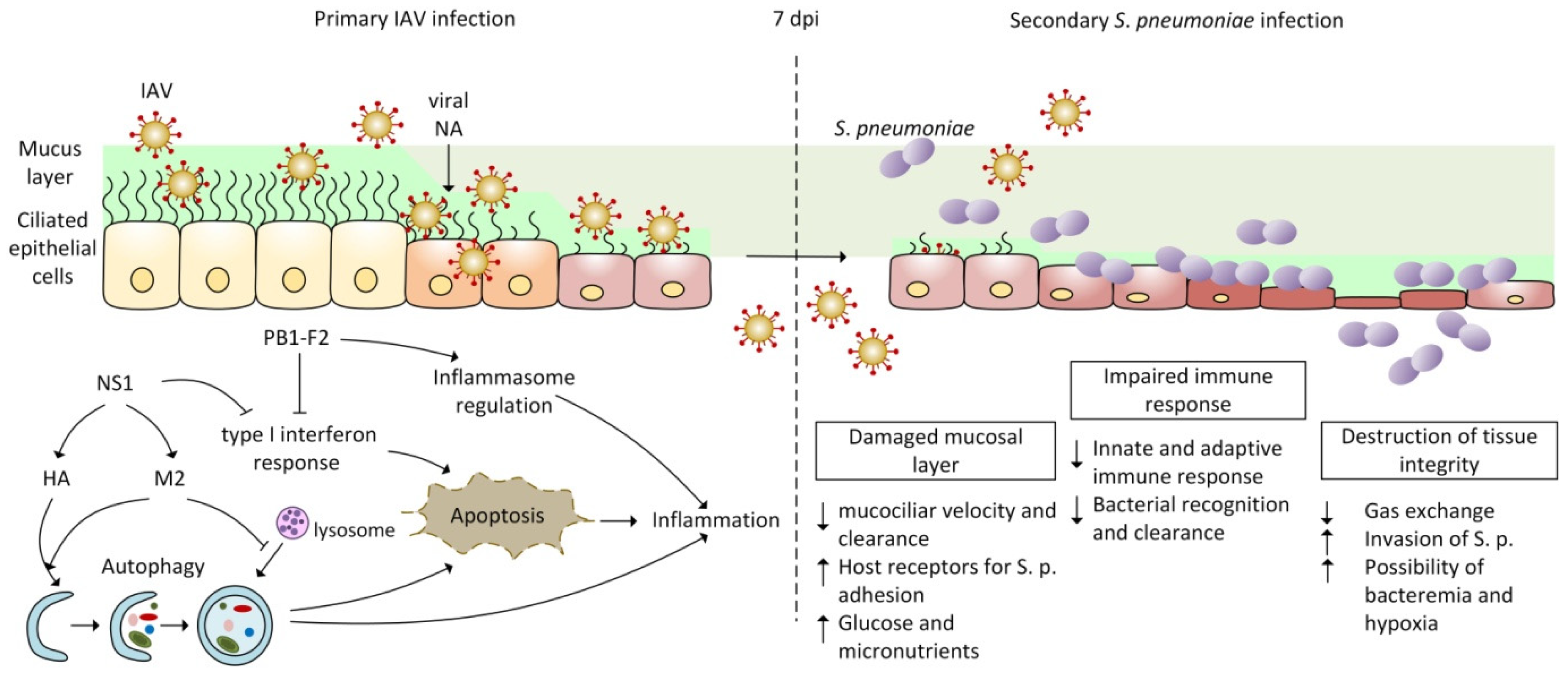
2.3. Co-Pathogenesis of IAV and Streptococcus pneumoniae
2.3.1. Disruption of Innate Immunity and Inflammatory Response during IAV and Bacterial Co-Infection
2.3.2. Autophagy and Apoptosis Mediated by Influenza Infection
3. Role of IAV Proteins in Viral and Bacterial Co-Infection
3.1. Characterization of PB1-F2 Protein
3.1.1. Apoptosis and Cytotoxicity Mediated by PB1-F2
3.1.2. PB1-F2 Pathogenic Markers Enhancing Secondary Bacterial Infection
3.2. Characterization of Hemagglutinin
3.2.1. Changes of HA Cleavage during Viral and Bacterial Co-Infection
3.2.2. The Role of Hemagglutinin in the Autophagy
3.3. Characterization of Neuraminidase
3.3.1. The Role of the Viral and Bacterial Neuraminidases in Co-Infection
3.3.2. Cooperation of NA and Galectins during Co-Infection
3.3.3. Impact of the Viral NA Activity on the Innate Immunity
3.4. Characterization of NS1
3.4.1. NS1 Interaction with Interferon Signaling Pathways Enhances the Development of Secondary Bacterial Infection
3.4.2. NS1 Motif Directly Involved in Co-Infection with S. pneumoniae
3.4.3. NS1 Manipulates Apoptosis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- The Top 10 Causes of Death. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 3 April 2022).
- Gavigan, P.; McCullers, J.A. Influenza: Annual seasonal severity. Curr. Opin. Pediatr. 2019, 1, 112–118. [Google Scholar] [CrossRef]
- Smith, A.M. Host-pathogen kinetics during influenza infection and coinfection: Insights from predictive modeling. Immunol. Rev. 2018, 285, 97–112. [Google Scholar] [CrossRef] [Green Version]
- Mehta, D.; Petes, C.; Gee, K.; Basta, S. The Role of Virus Infection in Deregulating the Cytokine Response to Secondary Bacterial Infection. J. Interferon Cytokine Res. 2015, 35, 925–934. [Google Scholar] [CrossRef]
- Metersky, M.L.; Masterton, R.G.; Lode, H.; File, T.M., Jr.; Babinchak, T. Epidemiology, microbiology, and treatment considerations for bacterial pneumonia complicating influenza. Int. J. Infect. Dis. 2012, 16, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Morris, D.E.; Cleary, D.W.; Clarke, S.C. Secondary Bacterial Infections Associated with Influenza Pandemics. Front. Microbiol. 2017, 8, 1041. [Google Scholar] [CrossRef] [Green Version]
- Jia, L.; Xie, J.; Zhao, J.; Cao, D.; Liang, Y.; Hou, X.; Wang, L.; Li, Z. Mechanisms of Severe Mortality-Associated Bacterial Co-infections Following Influenza Virus Infection. Front. Cell Infect. Microbiol. 2017, 7, 338. [Google Scholar] [CrossRef]
- Martin-Loeches, I.; van Someren Gréve, F.; Schultz, M.J. Bacterial pneumonia as an influenza complication. Curr. Opin. Infect. Dis. 2017, 30, 201–207. [Google Scholar] [CrossRef]
- Metzger, D.W.; Sun, K. Immune Dysfunction and Bacterial Co-Infections following Influenza. J. Immunol. 2013, 191, 2047–2052. [Google Scholar] [CrossRef] [Green Version]
- Jansen, A.G.; Sanders, E.A.; Van Der Ende, A.; Van Loon, A.M.; Hoes, A.W.; Hak, E. Invasive pneumococcal and meningococcal disease: Association with influenza virus and respiratory syncytial virus activity? Epidemiol. Infect. 2008, 136, 1448–1454. [Google Scholar] [CrossRef]
- Weiser, J.N.; Ferreira, D.M.; Paton, J.C. Streptococcus pneumoniae: Transmission, colonization and invasion. Nat. Rev. Microbiol. 2018, 16, 355–367. [Google Scholar] [CrossRef]
- Chertow, D.S.; Memoli, M.J. Bacterial Coinfection in Influenza. JAMA 2013, 309, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Briestenská, K.; Mikušová, M.; Tomčíková, K.; Kostolanský, F.; Varečková, E. Quantification of bacteria by in vivo bioluminescence imaging in comparison with standard spread plate method and reverse transcription quantitative PCR (RT-qPCR). Arch. Microbiol. 2021, 203, 4737–4742. [Google Scholar] [CrossRef]
- Seki, M.; Kosai, K.; Yanagihara, K.; Higashiyama, Y.; Kurihara, S.; Izumikawa, K.; Miyazaki, Y.; Hirakata, Y.; Tashiro, T.; Kohno, S. Disease Severity in Patients with Simultaneous Influenza and Bacterial Pneumonia. Intern. Med. J. 2007, 46, 953–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- COVID-19 & Antibiotic Resistance. Available online: https://www.cdc.gov/drugresistance/covid19.html (accessed on 3 April 2022).
- Mirzaei, R.; Goodarzi, P.; Asadi, M.; Soltani, A.; Aljanabi, H.; Jeda, A.S.; Dashtbin, S.; Jalalifar, S.; Mohammadzadeh, R.; Teimoori, A.; et al. Bacterial co-infections with SARS-CoV-2. IUBMB Life 2020, 72, 2097–2111. [Google Scholar] [CrossRef] [PubMed]
- Sender, V.; Hentrich, K.; Henriques-Normark, B. Virus-Induced Changes of the Respiratory Tract Environment Promote Secondary Infections With Streptococcus pneumoniae. Front. Cell. Infect. Microbiol. 2021, 11, 643326. [Google Scholar] [CrossRef]
- McCullers, J.A. Insights into the Interaction between Influenza Virus and Pneumococcus. Clin. Microbiol. Rev. 2006, 19, 571–582. [Google Scholar] [CrossRef] [Green Version]
- Morens, D.M.; Taubenberger, J.K.; Fauci, A.S. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: Implications for pandemic influenza preparedness. J. Infect. Dis. 2008, 198, 962–970. [Google Scholar] [CrossRef]
- Fislová, T.; Gocník, M.; Sládková, T.; Durmanová, V.; Rajcáni, J.; Varecková, E.; Mucha, V.; Kostolanský, F. Multiorgan distribution of human influenza A virus strains observed in a mouse model. Arch. Virol. 2009, 154, 409–419. [Google Scholar] [CrossRef]
- Kuiken, T.; Riteau, B.; Fouchier, R.A.M.; Rimmelzwaan, G.F. Pathogenesis of influenza virus infections: The good, the bad and the ugly. Curr. Opin. Virol. 2012, 2, 276–286. [Google Scholar] [CrossRef]
- Peteranderl, C.; Herold, S.; Schmoldt, C. Human Influenza Virus Infections. Semin. Respir. Crit. Care Med. 2016, 37, 487–500. [Google Scholar] [CrossRef]
- Francis, M.E.; King, M.L.; Kelvin, A.A. Back to the Future for Influenza Preimmunity—Looking Back at Influenza Virus History to Infer the Outcome of Future Infections. Viruses 2019, 11, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavares, L.P.; Teixeira, M.M.; Garcia, C.C. The inflamatory response triggered by Influenza virus: A two edged sword. Inflamm. Res. 2017, 66, 283–302. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, S.; Goraya, M.U.; Maarouf, M.; Huang, S.; Chen, J.L. Host Immune Response to Influenza A Virus Infection. Front. Immunol. 2018, 9, 320. [Google Scholar] [CrossRef] [Green Version]
- LeMessurier, K.S.; Tiwary, M.; Morin, N.P.; Samarasinghe, A.E. Respiratory Barrier as a Safeguard and Regulator of Defense Against Influenza A Virus and Streptococcus pneumoniae. Front. Immunol. 2020, 11, 3. [Google Scholar] [CrossRef] [Green Version]
- Shim, J.M.; Kim, J.; Tenson, T.; Min, J.Y.; Kainov, D.E. Influenza Virus Infection, Interferon Response, Viral Counter-Response, and Apoptosis. Viruses 2017, 9, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Metcalf, J.P. The Role of Type I IFNs in Influenza: Antiviral Superheroes or Immunopathogenic Villains? J. Innate Immun. 2020, 12, 437–447. [Google Scholar] [CrossRef]
- Nogales, A.; Aydillo, T.; Ávila-Pérez, G.; Escalera, A.; Chiem, K.; Cadagan, R.; Dediego, M.L.; Li, F.; García-Sastre, A.; Martínez-Sobrido, L. Functional Characterization and Direct Comparison of Influenza A, B, C, and D NS1 Proteins in vitro and in vivo. Front. Microbiol. 2019, 10, 2862. [Google Scholar] [CrossRef]
- Chen, W.H.; Toapanta, F.R.; Shirey, K.A.; Zhang, L.; Giannelou, A.; Page, C.; Frieman, M.B.; Vogel, S.N.; Cross, A.S. Potential role for alternatively activated macrophages in the secondary bacterial infection during recovery from influenza. Immunol. Lett. 2012, 141, 227–234. [Google Scholar] [CrossRef] [Green Version]
- Kudva, A.; Scheller, E.V.; Robinson, K.M.; Crowe, C.R.; Choi, S.M.; Slight, S.R.; Khader, S.A.; Dubin, P.J.; Enelow, R.I.; Kolls, J.K.; et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J. Immunol. 2011, 186, 1666–1674. [Google Scholar] [CrossRef] [Green Version]
- Shahangian, A.; Chow, E.K.; Tian, X.; Kang, J.R.; Ghaffari, A.; Liu, S.Y.; Belperio, J.A.; Cheng, G.; Deng, J.C. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J. Clin. Investig. 2009, 119, 1910–1920. [Google Scholar] [CrossRef]
- Shepardson, K.M.; Larson, K.; Morton, R.V.; Prigge, J.R.; Schmidt, E.E.; Huber, V.C.; Rynda-Apple, A. Differential Type I Interferon Signaling Is a Master Regulator of Susceptibility to Postinfluenza Bacterial Superinfection. MBio 2016, 7, e00506-16. [Google Scholar] [CrossRef] [Green Version]
- Techasaensiri, B.; Techasaensiri, C.; Mejias, A.; McCracken, G.H., Jr.; Ramilo, O. Viral coinfections in children with invasive pneumococcal disease. Pediatr. Infect. Dis. J. 2010, 29, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Hao, W.; Wang, L.; Li, S. Roles of the Non-Structural Proteins of Influenza A Virus. Pathogens 2020, 9, 812. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, E.C.; Charles, P.D.; Hester, S.S.; Thomas, B.; Trudgian, D.; Martínez-Alonso, M.; Fodor, E. Conserved and host-specific features of influenza virion architecture. Nat. Commun. 2014, 5, 4816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chlanda, P.; Zimmerberg, J. Protein-lipid interactions critical to replication of the influenza A virus. FEBS Lett. 2016, 590, 1940–1954. [Google Scholar] [CrossRef] [Green Version]
- Sriwilaijaroen, N.; Suzuki, Y. Molecular basis of the structure and function of H1 hemagglutinin of influenza virus. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2012, 88, 226–249. [Google Scholar] [CrossRef] [Green Version]
- Dou, D.; Revol, R.; Östbye, H.; Wang, H.; Daniels, R. Influenza A Virus Cell Entry, Replication, Virion Assembly and Movement. Front. Immunol. 2018, 9, 1581. [Google Scholar] [CrossRef]
- Li, S.; Sieben, C.; Ludwig, K.; Höfer, C.T.; Chiantia, S.; Herrmann, A.; Eghiaian, F.; Schaap, I.A. pH-Controlled two-step uncoating of influenza virus. Biophys. J. 2014, 106, 1447–1456. [Google Scholar] [CrossRef] [Green Version]
- Shaw, M.L.; Palese, P. Orthomyxoviridae: The Viruses and Their Replication. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P., Eds.; Lippincott Williams & Wilkins, A Wolters Kluwer: Philadelphia, PA, USA, 2013; Volume 40, pp. 1151–1185. ISBN 978-1-45-110563-6. [Google Scholar]
- Engel, D. The influenza virus NS1 protein as a therapeutic target. Antivir. Res. 2013, 99, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Hale, B.G.; Randall, R.E.; Ortín, J.; Jackson, D. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 2008, 89, 2359–2376. [Google Scholar] [CrossRef]
- Huang, S.; Chen, J.; Chen, Q.; Wang, H.; Yao, Y.; Chen, J.; Chen, Z. A second CRM1-dependent nuclear export signal in the influenza A virus NS2 protein contributes to the nuclear export of viral ribonucleoproteins. J. Virol. 2013, 87, 767–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paterson, D.; Fodor, E. Emerging Roles for the Influenza A Virus Nuclear Export Protein (NEP). PLoS Pathog. 2012, 8, e1003019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, J.S.; Mistry, B.; Haslam, S.M.; Barclay, W.S. Host and viral determinants of influenza A virus species specificity. Nat. Rev. Microbiol. 2018, 17, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Boianelli, A.; Nguyen, V.K.; Ebensen, T.; Schulze, K.; Wilk, E.; Sharma, N.; Stegemann-Koniszewski, S.; Bruder, D.; Toapanta, F.R.; Guzmán, C.A.; et al. Modeling Influenza Virus Infection: A Roadmap for Influenza Research. Viruses 2015, 7, 5274–5304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, L.; Nogales, A.; Martínez-Sobrido, L. Influenza A Virus Studies in a Mouse Model of Infection. JoVE 2017, 127, 55898. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, S.; Kawaoka, Y. The pathogenesis of influenza virus infections: The contributions of virus and host factors. Curr. Opin. Immunol. 2011, 23, 481–486. [Google Scholar] [CrossRef] [Green Version]
- Godlee, A.; Almond, M.H.; Dong, T. Pathogenesis of influenza: Virus–host interactions. Expert Rev. Anti Infect. Ther. 2011, 9, 573–575. [Google Scholar] [CrossRef]
- Kamal, R.P.; Alymova, I.V.; York, I.A. Evolution and Virulence of Influenza A Virus Protein PB1-F2. Int. J. Mol. Sci. 2017, 19, 96. [Google Scholar] [CrossRef] [Green Version]
- Košík, I.; Práznovská, M.; Košíková, M.; Bobišová, Z.; Hollý, J.; Varečková, E.; Kostolanský, F.; Russ, G. The Ubiquitination of the Influenza A Virus PB1-F2 Protein Is Crucial for Its Biological Function. PLoS ONE 2015, 10, e0118477. [Google Scholar] [CrossRef] [Green Version]
- Types of Influenza Viruses. Available online: https://www.cdc.gov/flu/about/viruses/types.htm (accessed on 3 April 2022).
- Bakaletz, L.O. Viral–bacterial co-infections in the respiratory tract. Curr. Opin. Microbiol. 2017, 35, 30–35. [Google Scholar] [CrossRef]
- Hanada, S.; Pirzadeh, M.; Carver, K.Y.; Deng, J.C. Respiratory Viral Infection-Induced Microbiome Alterations and Secondary Bacterial Pneumonia. Front. Immunol. 2018, 9, 2640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumpitsch, C.; Koskinen, K.; Schöpf, V.; Moissl-Eichinger, C. The microbiome of the upper respiratory tract in health and disease. BMC Biol. 2019, 17, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streptococcus Pneumoniae. Available online: https://www.cdc.gov/pneumococcal/clinicians/streptococcus-pneumoniae.html (accessed on 3 April 2022).
- Brooks, L.R.K.; Mias, G.I. Streptococcus pneumoniae’s Virulence and Host Immunity: Aging, Diagnostics, and Prevention. Front. Immunol. 2018, 9, 1366. [Google Scholar] [CrossRef] [PubMed]
- Kadioglu, A.; Weiser, J.N.; Paton, J.C.; Andrew, P.W. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat. Rev. Microbiol. 2008, 6, 288–301. [Google Scholar] [CrossRef]
- Paton, J.C.; Trappetti, C. Streptococcus pneumoniae Capsular Polysaccharide. Microbiol. Spectr. 2019, 7, 33. [Google Scholar] [CrossRef]
- Chao, Y.; Marks, L.R.; Pettigrew, M.M.; Hakansson, A.P. Streptococcus pneumoniae biofilm formation and dispersion during colonisation and disease. Front. Cell. Infect. Microbiol. 2015, 4, 194. [Google Scholar] [CrossRef] [Green Version]
- Domenech, M.; García, E.; Moscoso, M. Biofilm formation in Streptococcus pneumoniae. Microb. Biotechnol. 2012, 5, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Marks, L.R.; Reddinger, R.M.; Hakansson, A.P. High Levels of Genetic Recombination during Nasopharyngeal Carriage and Biofilm Formation in Streptococcus pneumoniae. MBio 2012, 3, e00200-12. [Google Scholar] [CrossRef] [Green Version]
- Shak, J.R.; Vidal, J.E.; Klugman, K.P. Influence of bacterial interactions on pneumococcal colonization of the nasopharynx. Trends Microbiol. 2013, 21, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Bogaert, D.; De Groot, R.; Hermans, P. Streptococcus pneumoniae colonisation: The key to pneumococcal disease. Lancet Infect. Dis. 2004, 4, 144–154. [Google Scholar] [CrossRef]
- Henriques-Normark, B.; Tuomanen, E.I. The Pneumococcus: Epidemiology, Microbiology, and Pathogenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a010215. [Google Scholar] [CrossRef] [PubMed]
- Yau, B.; Hunt, N.; Mitchell, A.; Too, L. Blood–Brain Barrier Pathology and CNS Outcomes in Streptococcus pneumoniae Meningitis. Int. J. Mol. Sci. 2018, 19, 3555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Egea, V.; Muñoz, P.; Valerio, M.; de Alarcón, A.; Lepe, J.A.; Miró, J.M.; Gálvez-Acebal, J.; García-Pavía, P.; Navas, E.; Goenaga, M.A.; et al. Characteristics and Outcome of Streptococcus pneumoniae Endocarditis in the XXI Century: A Systematic Review of 111 Cases (2000–2013). Medicine 2015, 94, e1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergenfelz, C.; Hakansson, A.P. Streptococcus pneumoniae Otitis Media Pathogenesis and How It Informs Our Understanding of Vaccine Strategies. Curr. Otorhinolaryngol. 2017, 5, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Peña, M.T.; Preciado, D.; Orestes, M.; Choi, S. Orbital complications of acute sinusitis: Changes in the post-pneumococcal vaccine era. JAMA Otolaryngol. Head Neck Surg. 2013, 139, 223–227. [Google Scholar] [CrossRef] [Green Version]
- Tinkelman, D.G.; Silk, H.J. Clinical and Bacteriologic Features of Chronic Sinusitis in Children. Am. J. Dis. Child. 1989, 143, 938–941. [Google Scholar] [CrossRef]
- Kim, P.E.; Musher, D.M.; Glezen, W.P.; Rodriguez-Barradas, M.C.; Nahm, W.K.; Wright, C.E. Association of Invasive Pneumococcal Disease with Season, Atmospheric Conditions, Air Pollution, and the Isolation of Respiratory Viruses. Clin. Infect. Dis. 1996, 22, 100–106. [Google Scholar] [CrossRef] [Green Version]
- Klugman, K.P.; Chien, Y.W.; Madhi, S.A. Pneumococcal pneumonia and influenza: A deadly combination. Vaccine 2009, 27, C9–C14. [Google Scholar] [CrossRef]
- Nair, H.; Brooks, W.A.; Katz, M.; Roca, A.; Berkley, J.A.; Madhi, S.A.; Simmerman, J.M.; Gordon, A.; Sato, M.; Howie, S.; et al. Global burden of respiratory infections due to seasonal influenza in young children: A systematic review and meta-analysis. Lancet 2011, 378, 1917–1930. [Google Scholar] [CrossRef] [Green Version]
- Rudd, J.M.; Ashar, H.K.; Chow, V.T.; Teluguakula, N. Lethal Synergism between Influenza and Streptococcus pneumoniae. J. Infect. Pulm. Dis. 2016, 2, 10-16966. [Google Scholar] [CrossRef]
- Pettigrew, M.M.; Marks, L.R.; Kong, Y.; Gent, J.F.; Roche-Hakansson, H.; Hakansson, A.P. Dynamic Changes in the Streptococcus pneumoniae Transcriptome during Transition from Biofilm Formation to Invasive Disease upon Influenza A Virus Infection. Infect. Immun. 2014, 82, 4607–4619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCullers, J.A.; Bartmess, K.C. Role of neuraminidase in lethal synergism between influenza virus and Streptococcus pneumoniae. J. Infect. Dis. 2003, 187, 1000–1009. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, E.R.; Lenz, L.L. Inflammation as a Modulator of Host Susceptibility to Pulmonary Influenza, Pneumococcal, and Co-Infections. Front. Immunol. 2020, 11, 105. [Google Scholar] [CrossRef] [Green Version]
- Brealey, J.C.; Sly, P.D.; Young, P.R.; Chappell, K.J. Viral bacterial co-infection of the respiratory tract during early childhood. FEMS Microbiol. Lett. 2015, 362, fnv062. [Google Scholar] [CrossRef] [Green Version]
- Gounder, A.P.; Boon, A.C.M. Influenza Pathogenesis: The Effect of Host Factors on Severity of Disease. J. Immunol. 2019, 202, 341–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCullers, J.A. The co-pathogenesis of influenza viruses with bacteria in the lung. Nat. Rev. Microbiol. 2014, 12, 252–262. [Google Scholar] [CrossRef]
- Kash, J.C.; Taubenberger, J.K. The Role of Viral, Host, and Secondary Bacterial Factors in Influenza Pathogenesis. Am. J. Pathol. 2015, 185, 1528–1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oggioni, M.R.; Trappetti, C.; Kadioglu, A.; Cassone, M.; Iannelli, F.; Ricci, S.; Andrew, P.W.; Pozzi, G. Switch from planktonic to sessile life: A major event in pneumococcal pathogenesis. Mol. Microbiol. 2006, 61, 1196–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taubenberger, J.K.; Kash, J.C. Insights on influenza pathogenesis from the grave. Virus Res. 2011, 162, 2–7. [Google Scholar] [CrossRef] [Green Version]
- Pittet, L.A.; Hall-Stoodley, L.; Rutkowski, M.R.; Harmsen, A.G. Influenza Virus Infection Decreases Tracheal Mucociliary Velocity and Clearance of Streptococcus pneumoniae. Am. J. Respir. Cell Mol. 2010, 42, 450–460. [Google Scholar] [CrossRef] [Green Version]
- Low, D. Reducing antibiotic use in influenza: Challenges and rewards. Clin. Microbiol. Infect. 2008, 14, 298–306. [Google Scholar] [CrossRef] [Green Version]
- Marks, L.R.; Davidson, B.A.; Knight, P.R.; Hakansson, A.P. Interkingdom Signaling Induces Streptococcus pneumoniae Biofilm Dispersion and Transition from Asymptomatic Colonization to Disease. MBio 2013, 4, e00438-13. [Google Scholar] [CrossRef] [Green Version]
- McNamee, L.A.; Harmsen, A.G. Both Influenza-Induced Neutrophil Dysfunction and Neutrophil-Independent Mechanisms Contribute to Increased Susceptibility to a Secondary Streptococcus pneumoniae Infection. Infect. Immun. 2006, 74, 6707–6721. [Google Scholar] [CrossRef] [Green Version]
- Ballinger, M.N.; Standiford, T.J. Postinfluenza bacterial pneumonia: Host defenses gone awry. J. Interferon Cytokine Res. 2010, 30, 643–652. [Google Scholar] [CrossRef] [Green Version]
- Ghoneim, H.E.; Thomas, P.G.; McCullers, J.A. Depletion of alveolar macrophages during influenza infection facilitates bacterial superinfections. J. Immunol. 2013, 191, 1250–1259. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Chen, C.J.; Mullarkey, C.E.; Hamilton, J.R.; Wong, C.K.; Leon, P.E.; Uccellini, M.B.; Chromikova, V.; Henry, C.; Hoffman, K.W.; et al. Alveolar macrophages are critical for broadly-reactive antibody-mediated protection against influenza A virus in mice. Nat. Commun. 2017, 8, 846. [Google Scholar] [CrossRef]
- Nicol, M.Q.; Dutia, B.M. The role of macrophages in influenza A virus infection. Future Virol. 2014, 9, 847–862. [Google Scholar] [CrossRef]
- Wilden, J.J.; Jacob, J.C.; Ehrhardt, C.; Ludwig, S.; Boergeling, Y. Altered Signal Transduction in the Immune Response to Influenza Virus and S. pneumoniae or S. aureus Co-Infections. Int. J. Mol. Sci. 2021, 22, 5486. [Google Scholar] [CrossRef] [PubMed]
- LeVine, A.M.; Koeningsknecht, V.; Stark, J.M. Decreased pulmonary clearance of S. pneumoniae following influenza A infection in mice. J. Virol. Methods 2001, 94, 173–186. [Google Scholar] [CrossRef]
- McCullers, J.A.; Rehg, J.E. Lethal synergism between influenza virus and Streptococcus pneumoniae: Characterization of a mouse model and the role of platelet-activating factor receptor. J. Infect. Dis. 2002, 186, 341–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.M.; Adler, F.R.; Ribeiro, R.M.; Gutenkunst, R.N.; McAuley, J.L.; McCullers, J.A.; Perelson, A.S. Kinetics of coinfection with influenza A virus and Streptococcus pneumoniae. PLoS Pathog. 2013, 9, e1003238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.M.; McCullers, J.A. Secondary Bacterial Infections in Influenza Virus Infection Pathogenesis. In Influenza Pathogenesis and Control—Volume I. Current Topics in Microbiology and Immunology; Compans, R., Oldstone, M., Eds.; Springer: Cham, Switzerland, 2014; Volume 385, pp. 327–356. ISBN 978-3-319-11155-1. [Google Scholar]
- Robinson, K.M.; Kolls, J.K.; Alcorn, J.F. The immunology of influenza virus-associated bacterial pneumonia. Curr. Opin. Immunol. 2015, 34, 59–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Didierlaurent, A.; Goulding, J.; Patel, S.; Snelgrove, R.; Low, L.; Bebien, M.; Lawrence, T.; van Rijt, L.S.; Lambrecht, B.N.; Sirard, J.C.; et al. Sustained desensitization to bacterial Toll-like receptor ligands after resolution of respiratory influenza infection. J. Exp. Med. 2008, 205, 323–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snelgrove, R.J.; Goulding, J.; Didierlaurent, A.M.; Lyonga, D.; Vekaria, S.; Edwards, L.; Gwyer, E.; Sedgwick, J.D.; Barclay, A.N.; Hussell, T. A critical function for CD200 in lung immune homeostasis and the severity of influenza infection. Nat. Immunol. 2008, 9, 1074–1083. [Google Scholar] [CrossRef]
- Li, W.; Moltedo, B.; Moran, T.M. Type I interferon induction during influenza virus infection increases susceptibility to secondary Streptococcus pneumoniae infection by negative regulation of γδ T cells. J. Virol. 2012, 86, 12304–12312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, K.; Metzger, D.W. Inhibition of pulmonary antibacterial defense by interferon-gamma during recovery from influenza infection. Nat. Med. 2008, 14, 558–564. [Google Scholar] [CrossRef]
- Branchett, W.J.; Lloyd, C.M. Regulatory cytokine function in the respiratory tract. Mucosal Immunol. 2019, 12, 589–600. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Madan, R.; Karp, C.L.; Braciale, T.J. Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nat. Med. 2009, 15, 277–284. [Google Scholar] [CrossRef]
- Camp, J.V.; Jonsson, C.B. A Role for Neutrophils in Viral Respiratory Disease. Front. Immunol. 2017, 8, 550. [Google Scholar] [CrossRef] [Green Version]
- Colamussi, M.L.; White, M.R.; Crouch, E.; Hartshorn, K.L. Influenza A virus accelerates neutrophil apoptosis and markedly potentiates apoptotic effects of bacteria. Blood 1999, 93, 2395–2403. [Google Scholar] [CrossRef]
- Fernandez, M.V.; Miller, E.; Krammer, F.; Gopal, R.; Greenbaum, B.D.; Bhardwaj, N. Ion efflux and influenza infection trigger NLRP3 inflammasome signaling in human dendritic cells. J. Leukoc. Biol. 2016, 99, 723–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, H.; Liu, Y.; Sia, S.F.; Peiris, J.S.M.; Lau, Y.L.; Tu, W. Avian influenza virus directly infects human natural killer cells and inhibits cell activity. Virol. Sin. 2017, 32, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Frank, K.; Paust, S. Dynamic Natural Killer Cell and T Cell Responses to Influenza Infection. Front. Cell. Infect. Microbiol. 2020, 10, 425. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zheng, J.; Liu, Y.; Wen, L.; Huang, L.; Xiang, Z.; Lam, K.T.; Lv, A.; Mao, H.; Lau, Y.L.; et al. Uncompromised NK cell activation is essential for virus-specific CTL activity during acute influenza virus infection. Cell. Mol. Immunol. 2018, 15, 827–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz-Cherry, S. Viral Interference: The Case of Influenza Viruses. J. Infect. Dis. 2015, 212, 1690–1691. [Google Scholar] [CrossRef] [Green Version]
- Small, C.L.; Shaler, C.R.; McCormick, S.; Jeyanathan, M.; Damjanovic, D.; Brown, E.G.; Arck, P.; Jordana, M.; Kaushic, C.; Ashkar, A.A.; et al. Influenza infection leads to increased susceptibility to subsequent bacterial superinfection by impairing NK cell responses in the lung. J. Immunol. 2010, 184, 2048–2056. [Google Scholar] [CrossRef] [Green Version]
- Hinshaw, V.S.; Olsen, C.W.; Dybdahl-Sissoko, N.; Evans, D. Apoptosis: A mechanism of cell killing by influenza A and B viruses. J. Virol. 1994, 68, 3667–3673. [Google Scholar] [CrossRef] [Green Version]
- Mehrbod, P.; Ande, S.R.; Alizadeh, J.; Rahimizadeh, S.; Shariati, A.; Malek, H.; Hashemi, M.; Glover, K.K.M.; Sher, A.A.; Coombs, K.M.; et al. The roles of apoptosis, autophagy and unfolded protein response in arbovirus, influenza virus, and HIV infections. Virulence 2019, 10, 376–413. [Google Scholar] [CrossRef] [Green Version]
- Morris, S.J.; Price, G.E.; Barnett, J.M.; Hiscox, S.A.; Smith, H.; Sweet, C. Role of neuraminidase in influenza virus-induced apoptosis. J. Gen. Virol. 1999, 80, 137–146. [Google Scholar] [CrossRef]
- Zhou, A.; Zhang, W.; Dong, X.; Liu, M.; Chen, H.; Tang, B. The battle for autophagy between host and influenza A virus. Virulence 2021, 13, 46–59. [Google Scholar] [CrossRef]
- Atkin-Smith, G.K.; Duan, M.; Chen, W.; Poon, I.K.H. The induction and consequences of Influenza A virus-induced cell death. Cell Death Dis. 2018, 9, 1002. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Yang, Y.; Wang, H.; Luo, J.; Huang, X.; You, J.; Wang, B.; Li, M. Role of Autophagy and Apoptosis in the Postinfluenza Bacterial Pneumonia. Biomed. Res. Int. 2016, 2016, 3801026. [Google Scholar] [CrossRef] [Green Version]
- Zhirnov, O.P.; Klenk, H.D. Influenza A virus proteins NS1 and hemagglutinin along with M2 are involved in stimulation of autophagy in infected cells. J. Virol. 2013, 87, 13107–13114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhirnov, O.P.; Konakova, T.E.; Wolff, T.; Klenk, H.D. NS1 protein of influenza A virus down-regulates apoptosis. J. Virol. 2002, 76, 1617–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, A.H.; Lee, D.C.; Leon, T.Y.; Lau, A.S. Role for autophagy in cellular response to influenza virus infection. Hong Kong Med. J. 2014, 20, 20–24. [Google Scholar]
- Thulasi Raman, S.N.; Zhou, Y. Networks of Host Factors that Interact with NS1 Protein of Influenza A Virus. Front. Microbiol. 2016, 7, 654. [Google Scholar] [CrossRef] [Green Version]
- Yordy, B.; Iwasaki, A. Autophagy in the control and pathogenesis of viral infection. Curr. Opin. Virol. 2011, 1, 196–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Jiang, X.; Liu, D.; Fan, Z.; Hu, X.; Yan, J.; Wang, M.; Gao, G.F. Autophagy is involved in influenza A virus replication. Autophagy 2009, 5, 321–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, W.; Yeung, A.C.; Chan, P.K. Apoptosis, cytokine and chemokine induction by non-structural 1 (NS1) proteins encoded by different influenza subtypes. Virol. J. 2011, 8, 554. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Sun, Q.; Mi, R.; Zhang, H. Avian influenza A virus H5N1 causes autophagy-mediated cell death through suppression of mTOR signaling. J. Genet. Genom. 2011, 38, 533–537. [Google Scholar] [CrossRef]
- Sun, Y.; Li, C.; Shu, Y.; Ju, X.; Zou, Z.; Wang, H.; Rao, S.; Guo, F.; Liu, H.; Nan, W.; et al. Inhibition of autophagy ameliorates acute lung injury caused by avian influenza A H5N1 infection. Sci. Signal. 2012, 5, ra16. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhu, Y.; Zhao, J.; Ren, C.; Li, P.; Chen, H.; Jin, M.; Zhou, H. Autophagy Promotes Replication of Influenza A Virus In Vitro. J. Virol. 2019, 93, e01984-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gannagé, M.; Dormann, D.; Albrecht, R.; Dengjel, J.; Torossi, T.; Rämer, P.C.; Lee, M.; Strowig, T.; Arrey, F.; Conenello, G.; et al. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 2009, 6, 367–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herold, S.; Ludwig, S.; Pleschka, S.; Wolff, T. Apoptosis signaling in influenza virus propagation, innate host defense, and lung injury. J. Leukoc. Biol. 2012, 92, 75–82. [Google Scholar] [CrossRef]
- Iwai, A.; Shiozaki, T.; Miyazaki, T. Relevance of signaling molecules for apoptosis induction on influenza A virus replication. Biochem. Biophys. Res. Commun. 2013, 441, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Schultz-Cherry, S.; Dybdahl-Sissoko, N.; Neumann, G.; Kawaoka, Y.; Hinshaw, V.S. Influenza Virus Ns1 Protein Induces Apoptosis in Cultured Cells. J. Virol. 2001, 75, 7875–7881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Calvo, P.A.; Malide, D.; Gibbs, J.; Schubert, U.; Bacik, I.; Basta, S.; O’Neill, R.; Schickli, J.; Palese, P.; et al. A novel influenza A virus mitochondrial protein that induces cell death. Nat. Med. 2001, 7, 1306–1312. [Google Scholar] [CrossRef]
- Yeganeh, B.; Ghavami, S.; Rahim, M.N.; Klonisch, T.; Halayko, A.J.; Coombs, K.M. Autophagy activation is required for influenza A virus-induced apoptosis and replication. Biochim. Biophys. Acta—Mol. Cell Res. 2018, 1865, 364–378. [Google Scholar] [CrossRef]
- Zhang, R.; Chi, X.; Wang, S.; Qi, B.; Yu, X.; Chen, J.L. The regulation of autophagy by influenza A virus. Biomed. Res. Int. 2014, 2014, 498083. [Google Scholar] [CrossRef]
- Kosai, K.; Seki, M.; Tanaka, A.; Morinaga, Y.; Imamura, Y.; Izumikawa, K.; Kakeya, H.; Yamamoto, Y.; Yanagihara, K.; Tomono, K.; et al. Increase of apoptosis in a murine model for severe pneumococcal pneumonia during influenza A virus infection. Jpn. J. Infect. Dis. 2011, 64, 451–457. [Google Scholar] [CrossRef]
- Cauley, L.S.; Vella, A.T. Why is coinfection with influenza virus and bacteria so difficult to control? Discov. Med. 2015, 19, 33–40. [Google Scholar]
- Feng, C.; Zhang, L.; Nguyen, C.; Vogel, S.N.; Goldblum, S.E.; Blackwelder, W.C.; Cross, A.S. Neuraminidase reprograms lung tissue and potentiates lipopolysaccharide-induced acute lung injury in mice. J. Immunol. 2013, 191, 4828–4837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, S.J.; Roche, A.M.; Weiser, J.N. Influenza promotes pneumococcal growth during coinfection by providing host sialylated substrates as a nutrient source. Cell Host Microbe 2014, 16, 55–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billharz, R.; Zeng, H.; Proll, S.C.; Korth, M.J.; Lederer, S.; Albrecht, R.; Goodman, A.G.; Rosenzweig, E.; Tumpey, T.M.; García-Sastre, A.; et al. The NS1 protein of the 1918 pandemic influenza virus blocks host interferon and lipid metabolism pathways. J Virol. 2009, 83, 10557–10570. [Google Scholar] [CrossRef] [Green Version]
- Ji, Z.X.; Wang, X.Q.; Liu, X.F. NS1: A Key Protein in the “Game” Between Influenza A Virus and Host in Innate Immunity. Front. Cell Infect. Microbiol. 2021, 11, 670177. [Google Scholar] [CrossRef]
- Halder, U.C.; Bagchi, P.; Chattopadhyay, S.; Dutta, D.; Chawla-Sarkar, M. Cell death regulation during influenza A virus infection by matrix (M1) protein: A model of viral control over the cellular survival pathway. Cell Death Dis. 2011, 2, e197. [Google Scholar] [CrossRef] [Green Version]
- Mayank, A.K.; Sharma, S.; Nailwal, H.; Lal, S.K. Nucleoprotein of influenza A virus negatively impacts antiapoptotic protein API5 to enhance E2F1-dependent apoptosis and virus replication. Cell Death Dis. 2015, 6, e2018. [Google Scholar] [CrossRef]
- Tripathi, S.; Batra, J.; Cao, W.; Sharma, K.; Patel, J.R.; Ranjan, P.; Kumar, A.; Katz, J.M.; Cox, N.J.; Lal, R.B.; et al. Influenza A virus nucleoprotein induces apoptosis in human airway epithelial cells: Implications of a novel interaction between nucleoprotein and host protein Clusterin. Cell Death Dis. 2013, 4, 562. [Google Scholar] [CrossRef] [Green Version]
- Ichinohe, T.; Pang, I.K.; Iwasaki, A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol. 2010, 11, 404–410. [Google Scholar] [CrossRef]
- Cheung, P.H.; Lee, T.T.; Chan, C.P.; Jin, D.Y. Influenza A virus PB1-F2 protein: An ambivalent innate immune modulator and virulence factor. J. Leukoc. Biol. 2020, 107, 763–771. [Google Scholar] [CrossRef]
- Laghlali, G.; Lawlor, K.E.; Tate, M.D. Die Another Way: Interplay between Influenza A Virus, Inflammation and Cell Death. Viruses 2020, 12, 401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conenello, G.M.; Tisoncik, J.R.; Rosenzweig, E.; Varga, Z.T.; Palese, P.; Katze, M.G. A single N66S mutation in the PB1-F2 protein of influenza A virus increases virulence by inhibiting the early interferon response in vivo. J. Virol. 2011, 85, 652–662. [Google Scholar] [CrossRef] [Green Version]
- Varga, Z.T.; Ramos, I.; Hai, R.; Schmolke, M.; García-Sastre, A.; Fernandez-Sesma, A.; Palese, P. The influenza virus protein PB1-F2 inhibits the induction of type I interferon at the level of the MAVS adaptor protein. PLoS Pathog. 2011, 7, e1002067. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Garcia-Sastre, A.; Xiao, X.; Wang, R.; Palese, P. Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog. 2005, 1, e4. [Google Scholar] [CrossRef]
- Wang, R.; Zhu, Y.; Ren, C.; Yang, S.; Tian, S.; Chen, H.; Jin, M.; Zhou, H. Influenza A virus protein PB1-F2 impairs innate immunity by inducing mitophagy. Autophagy 2021, 17, 496–511. [Google Scholar] [CrossRef] [PubMed]
- Alymova, I.V.; Samarasinghe, A.; Vogel, P.; Green, A.M.; Weinlich, R.; McCullers, J.A. A novel cytotoxic sequence contributes to influenza A viral protein PB1-F2 pathogenicity and predisposition to secondary bacterial infection. J. Virol. 2014, 88, 503–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alymova, I.V.; Green, A.M.; van de Velde, N.; McAuley, J.L.; Boyd, K.L.; Ghoneim, H.E.; McCullers, J.A. Immunopathogenic and antibacterial effects of H3N2 influenza A virus PB1-F2 map to amino acid residues 62, 75, 79, and 82. J. Virol. 2011, 85, 12324–12333. [Google Scholar] [CrossRef] [Green Version]
- McAuley, J.; Deng, Y.M.; Gilbertson, B.; Mackenzie-Kludas, C.; Barr, I.; Brown, L. Rapid evolution of the PB1-F2 virulence protein expressed by human seasonal H3N2 influenza viruses reduces inflammatory responses to infection. Virol. J. 2017, 14, 162. [Google Scholar] [CrossRef] [Green Version]
- Conenello, G.M.; Zamarin, D.; Perrone, L.A.; Tumpey, T.; Palese, P. A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog. 2007, 3, 1414–1421. [Google Scholar] [CrossRef]
- McAuley, J.L.; Tate, M.D.; MacKenzie-Kludas, C.J.; Pinar, A.; Zeng, W.; Stutz, A.; Latz, E.; Brown, L.E.; Mansell, A. Activation of the NLRP3 inflammasome by IAV virulence protein PB1-F2 contributes to severe pathophysiology and disease. PLoS Pathog. 2013, 9, e1003392. [Google Scholar] [CrossRef] [Green Version]
- Pinar, A.; Dowling, J.K.; Bitto, N.J.; Robertson, A.A.; Latz, E.; Stewart, C.R.; Drummond, G.R.; Cooper, M.A.; McAuley, J.L.; Tate, M.D.; et al. PB1-F2 Peptide Derived from Avian Influenza A Virus H7N9 Induces Inflammation via Activation of the NLRP3 Inflammasome. J. Biol. Chem. 2017, 292, 826–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshizumi, T.; Ichinohe, T.; Sasaki, O.; Otera, H.; Kawabata, S.; Mihara, K.; Koshiba, T. Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat. Commun. 2014, 5, 4713. [Google Scholar] [CrossRef]
- Wang, X.; Zheng, T.; Lin, L.; Zhang, Y.; Peng, X.; Yan, Y.; Lei, J.; Zhou, J.; Hu, B. Influenza A Virus Induces Autophagy By Its Hemagglutinin Binding To Cell Surface Heat Shock Protein 90aa1. Front. Microbiol. 2020, 11, 566348. [Google Scholar] [CrossRef]
- Klonoski, J.M.; Watson, T.; Bickett, T.E.; Svendsen, J.M.; Gau, T.J.; Britt, A.; Nelson, J.T.; Schlenker, E.H.; Chaussee, M.S.; Rynda-Apple, A.; et al. Contributions of Influenza Virus Hemagglutinin and Host Immune Responses Toward the Severity of Influenza Virus: Streptococcus pyogenes Superinfections. Viral immunol. 2018, 31, 457–469. [Google Scholar] [CrossRef]
- Nita-Lazar, M.; Banerjee, A.; Feng, C.; Amin, M.N.; Frieman, M.B.; Chen, W.H.; Cross, A.S.; Wang, L.X.; Vasta, G.R. Desialylation of airway epithelial cells during influenza virus infection enhances pneumococcal adhesion via galectin binding. Mol. Immunol. 2015, 65, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nita-Lazar, M.; Banerjee, A.; Feng, C.; Vasta, G.R. Galectins regulate the inflammatory response in airway epithelial cells exposed to microbial neuraminidase by modulating the expression of SOCS1 and RIG1. Mol. Immunol. 2015, 68, 194–202. [Google Scholar] [CrossRef] [Green Version]
- Fitzpatrick, D.R.; Bielefeldt-Ohmann, H. Transforming growth factor beta in infectious disease: Always there for the host and the pathogen. Trends Microbiol. 1999, 7, 232–236. [Google Scholar] [CrossRef]
- Li, N.; Ren, A.; Wang, X.; Fan, X.; Zhao, Y.; Gao, G.F.; Cleary, P.; Wang, B. Influenza viral neuraminidase primes bacterial coinfection through TGF-β-mediated expression of host cell receptors. Proc. Natl. Acad. Sci. USA 2015, 112, 238–243. [Google Scholar] [CrossRef] [Green Version]
- Dutta, A.; Huang, C.T.; Chen, T.C.; Lin, C.Y.; Chiu, C.H.; Lin, Y.C.; Chang, C.S.; He, Y.C. IL-10 inhibits neuraminidase-activated TGF-β and facilitates Th1 phenotype during early phase of infection. Nat. Commun. 2015, 6, 6374. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.S.; Huang, I.C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; García-Sastre, A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 2009, 5, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Rajsbaum, R.; Albrecht, R.A.; Wang, M.K.; Maharaj, N.P.; Versteeg, G.A.; Nistal-Villán, E.; García-Sastre, A.; Gack, M.U. Species-specific inhibition of RIG-I ubiquitination and IFN induction by the influenza A virus NS1 protein. PLoS Pathog. 2012, 8, e1003059. [Google Scholar] [CrossRef] [PubMed]
- Hatada, E.; Fukuda, R. Binding of influenza A virus NS1 protein to dsRNA in vitro. J. Gen. Virol. 1992, 73, 3325–3329. [Google Scholar] [CrossRef]
- Hatada, E.; Saito, S.; Fukuda, R. Mutant influenza viruses with a defective NS1 protein cannot block the activation of PKR in infected cells. J. Virol. 1999, 73, 2425–2433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmann, M.; Garcia-Sastre, A.; Carnero, E.; Pehamberger, H.; Wolff, K.; Palese, P.; Muster, T. Influenza virus NS1 protein counteracts PKR-mediated inhibition of replication. J. Virol. 2000, 74, 6203–6206. [Google Scholar] [CrossRef] [Green Version]
- Min, J.Y.; Li, S.; Sen, G.C.; Krug, R.M. A Site on the Influenza A Virus NS1 Protein Mediates Both Inhibition of PKR Activation and Temporal Regulation of Viral RNA Synthesis. Virology 2007, 363, 236–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, J.Y.; Krug, R.M. The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: Inhibiting the 2′-5′ oligo (A) synthe-tase/RNase L pathway. Proc. Natl. Acad. Sci. USA 2006, 103, 7100–7105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talon, J.; Horvath, C.M.; Polley, R.; Basler, C.F.; Muster, T.; Palese, P.; García-Sastre, A. Activation of Interferon Regulatory Factor 3 Is Inhibited by the Influenza A Virus NS1 Protein. J. Virol. 2000, 74, 7989–7996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, W.; Wei, X.; Guo, K.; Li, Y.; Lin, X.; Zou, Z.; Zhou, H.; Jin, M. The C-Terminal Effector Domain of Non-Structural Protein 1 of Influenza A Virus Blocks IFN-β Production by Targeting TNF Receptor-Associated Factor 3. Front. Immunol. 2017, 8, 779. [Google Scholar] [CrossRef]
- Feng, W.; Sun, X.; Shi, N.; Zhang, M.; Guan, Z.; Duan, M. Influenza a virus NS1 protein induced A20 contributes to viral replication by suppressing interferon-induced antiviral response. Biochem. Biophys. Res. Commun. 2017, 482, 1107–1113. [Google Scholar] [CrossRef]
- Jackson, D.; Hossain, M.J.; Hickman, D.; Perez, D.R.; Lamb, R.A. A new influenza virus virulence determinant: The NS1 protein four C-terminal residues modulate pathogenicity. Proc. Natl. Acad. Sci. USA 2008, 105, 4381–4386. [Google Scholar] [CrossRef] [Green Version]
- Hale, B.G.; Jackson, D.; Chen, Y.H.; Lamb, R.A.; Randall, R.E. Influenza A virus NS1 protein binds p85beta and activates phosphatidylinositol-3-kinase signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 14194–14199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, Y.K.; Liu, Q.; Tikoo, S.K.; Babiuk, L.A.; Zhou, Y. Influenza A virus NS1 protein activates the phosphatidylinositol 3-kinase (PI3K)/Akt pathway by direct interaction with the p85 subunit of PI3K. J. Gen. Virol. 2007, 88, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.K.; Li, Y.; Liu, Q.; Anderson, D.H.; Babiuk, L.A.; Zhou, Y. SH3 binding motif 1 in influenza A virus NS1 protein is essential for PI3K/Akt signaling pathway activation. J. Virol. 2007, 81, 12730–12739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, S.; Majumdar, S.; Vipat, V.C.; Mishra, A.C.; Chakrabarti, A.K. Non Structural Protein of Avian Influenza A (H11N1) Virus Is a Weaker Suppressor of Immune Responses But Capable of Inducing Apoptosis in Host Cells. Virol. J. 2012, 9, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Yang, Y.; Zhou, X.; Yang, Z.; Liu, X.; Cao, Z.; Song, H.; He, Y.; Huang, P. The NS1 protein of influenza A virus interacts with heat shock protein Hsp90 in human alveolar basal epithelial cells: Implication for virus-induced apoptosis. Virol. J. 2011, 8, 181. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Shen, Y.; Qiu, Y.; Shi, Z.; Shao, D.; Chen, P.; Tong, G.; Ma, Z. The non-Structural (NS1) Protein of Influenza A Virus Associates With p53 and Inhibits p53-mediated Transcriptional Activity and Apoptosis. Biochem. Biophys. Res. Commun. 2010, 395, 141–145. [Google Scholar] [CrossRef]
- Yan, Y.; Du, Y.; Wang, G.; Li, K. Non-structural protein 1 of H3N2 influenza A virus induces nucleolar stress via interaction with nucleolin. Sci. Rep. 2017, 7, 17761. [Google Scholar] [CrossRef] [Green Version]
- Zhirnov, O.P.; Klenk, H.D. Control of apoptosis in influenza virus-infected cells by up-regulation of Akt and p53 signaling. Apoptosis 2007, 12, 1419–1432. [Google Scholar] [CrossRef]
- Martin-Sancho, L.; Tripathi, S.; Rodriguez-Frandsen, A.; Pache, L.; Sanchez-Aparicio, M.; McGregor, M.J.; Haas, K.M.; Swaney, D.L.; Nguyen, T.T.; Mamede, J.I.; et al. Restriction factor compendium for influenza A virus reveals a mechanism for evasion of autophagy. Nat. Microbiol. 2021, 6, 1319–1333. [Google Scholar] [CrossRef]
- Beale, R.; Wise, H.; Stuart, A.; Ravenhill, B.J.; Digard, P.; Randow, F. A LC3-interacting motif in the influenza A virus M2 protein is required to subvert autophagy and maintain virion stability. Cell Host Microbe 2014, 15, 239–247. [Google Scholar] [CrossRef] [Green Version]
- Alymova, I.V.; York, I.A.; McCullers, J.A. Non-avian animal reservoirs present a source of influenza A PB1-F2 proteins with novel virulence-enhancing markers. PLoS ONE 2014, 9, e111603. [Google Scholar] [CrossRef] [PubMed]
- Alymova, I.V.; McCullers, J.A.; Kamal, R.P.; Vogel, P.; Green, A.M.; Gansebom, S.; York, I.A. Virulent PB1-F2 residues: Effects on fitness of H1N1 influenza A virus in mice and changes during evolution of human influenza A viruses. Sci. Rep. 2018, 8, 7474. [Google Scholar] [CrossRef]
- Bruns, K.; Studtrucker, N.; Sharma, A.; Fossen, T.; Mitzner, D.; Eissmann, A.; Tessmer, U.; Roder, R.; Henklein, P.; Wray, V.; et al. Structural characterization and oligomerization of PB1-F2, a proapoptotic influenza A virus protein. J. Biol. Chem. 2007, 282, 353–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevalier, C.; Al Bazzal, A.; Vidic, J.; Février, V.; Bourdieu, C.; Bouguyon, E.; Le Goffic, R.; Vautherot, J.F.; Bernard, J.; Moudjou, M.; et al. PB1-F2 influenza A virus protein adopts a beta-sheet conformation and forms amyloid fibers in membrane environments. J. Biol. Chem. 2010, 285, 13233–13243. [Google Scholar] [CrossRef] [Green Version]
- Henklein, P.; Bruns, K.; Nimtz, M.; Wray, V.; Tessmer, U.; Schubert, U. Influenza A virus protein PB1-F2: Synthesis and characterization of the biologically active full length protein and related peptides. J. Peptide Sci. 2005, 11, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Mazur, I.; Anhlan, D.; Mitzner, D.; Wixler, L.; Schubert, U.; Ludwig, S. The proapoptotic influenza A virus protein PB1-F2 regulates viral polymerase activity by interaction with the PB1 protein. Cell. Microbiol. 2008, 10, 1140–1152. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Chen, G.W.; Wang, C.H.; Huang, C.H.; Wang, Y.C.; Shih, S.R. Differential localization and function of PB1-F2 derived from different strains of influenza A virus. J. Virol. 2010, 84, 10051–10062. [Google Scholar] [CrossRef] [Green Version]
- Kosik, I.; Krejnusova, I.; Bystricka, M.; Polakova, K.; Russ, G. N-terminal region of the PB1-F2 protein is responsible for increased expression of influenza a viral protein PB1. Acta Virol. 2011, 55, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, A.K.; Pasricha, G. An insight into the PB1F2 protein and its multifunctional role in enhancing the pathogenicity of the influenza A viruses. Virology 2013, 440, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.M.; Adler, F.R.; McAuley, J.L.; Gutenkunst, R.N.; Ribeiro, R.M.; McCullers, J.A.; Perelson, A.S. Effect of 1918 pb1-f2 expression on influenza a virus infection kinetics. PLoS Comput. Biol. 2011, 7, e1001081. [Google Scholar] [CrossRef] [Green Version]
- Coleman, J.R. The PB1-F2 protein of Influenza A virus: Increasing pathogenicity by disrupting alveolar macrophages. Virol. J. 2007, 4, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, P.H.; Bird, N.; Mackenzie-Kludas, C.; Mansell, A.; Kedzierska, K.; Brown, L.; McAuley, J. Induction of memory cytotoxic T cells to influenza A virus and subsequent viral clearance is not modulated by PB1-F2-dependent inflammasome activation. Immunol. Cell. Biol. 2016, 94, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, J.S.; Malide, D.; Hornung, F.; Bennink, J.R.; Yewdell, J.W. The Influenza A Virus PB1-F2 Protein Targets the Inner Mitochondrial Membrane via a Predicted Basic Amphipathic Helix That Disrupts Mitochondrial Function. J. Virol. 2003, 77, 7214–7224. [Google Scholar] [CrossRef] [Green Version]
- Yamada, H.; Chounan, R.; Higashi, Y.; Kurihara, N.; Kido, H. Mitochondrial targeting sequence of the influenza A virus PB1-F2 protein and its function in mitochondria. FEBS Lett. 2004, 578, 331–336. [Google Scholar] [CrossRef] [Green Version]
- Varga, Z.T.; Palese, P. The influenza A virus protein PB1-F2. Virulence 2011, 2, 542–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAuley, J.L.; Hornung, F.; Boyd, K.L.; Smith, A.M.; McKeon, R.; Bennink, J.; Yewdell, J.W.; McCullers, J.A. Expression of the 1918 influenza A virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe 2007, 2, 240–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weeks-Gorospe, J.N.; Hurtig, H.R.; Iverson, A.R.; Schuneman, M.J.; Webby, R.J.; McCullers, J.A.; Huber, V.C. Naturally occurring swine influenza A virus PB1-F2 phenotypes that contribute to superinfection with Gram-positive respiratory pathogens. J. Virol. 2012, 86, 9035–9043. [Google Scholar] [CrossRef] [Green Version]
- Tate, M.D.; Mansell, A. An update on the NLRP3 inflammasome and influenza: The road to redemption or perdition? Curr. Opin. Immunol. 2018, 54, 80–85. [Google Scholar] [CrossRef]
- Zhao, C.; Zhao, W. NLRP3Inflammasome—A Key Player in Antiviral Responses. Front. Immunol. 2020, 11, 211. [Google Scholar] [CrossRef] [Green Version]
- Broz, P.; Monack, D.M. Molecular mechanisms of inflammasome activation during microbial infections. Immunol. Rev. 2011, 243, 174–190. [Google Scholar] [CrossRef]
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 2009, 30, 556–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guarda, G.; Braun, M.; Staehli, F.; Tardivel, A.; Mattmann, C.; Förster, I.; Farlik, M.; Decker, T.; Du Pasquier, R.A.; Romero, P.; et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 2011, 34, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Solbak, S.M.; Sharma, A.; Bruns, K.; Röder, R.; Mitzner, D.; Hahn, F.; Niebert, R.; Vedeler, A.; Henklein, P.; Henklein, P.; et al. Influenza A virus protein PB1-F2 from different strains shows distinct structural signatures. Biochim. Biophys. Acta 2013, 1834, 568–582. [Google Scholar] [CrossRef] [PubMed]
- Košík, I.; Hollý, J.; Russ, G. PB1-F2 expedition from the whole protein through the domain to aa residue function. Acta Virol. 2013, 57, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Staneková, Z.; Varečková, E. Conserved epitopes of influenza A virus inducing protective immunity and their prospects for universal vaccine development. Virol. J. 2010, 7, 351. [Google Scholar] [CrossRef] [Green Version]
- Tomčíková, K.; Varečková, E. Different mechanisms of the protection against influenza A infection mediated by broadly reactive HA2-specific antibodies. Acta Virol. 2019, 63, 347–365. [Google Scholar] [CrossRef]
- Varečková, E.; Mucha, V.; Kostolanský, F. HA2 glycopolypeptide of influenza A virus and antiviral immunity. Acta Virol. 2013, 57, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Steinhauer, D.A. Role of Hemagglutinin Cleavage for the Pathogenicity of Influenza Virus. Virology 1999, 258, 1–20. [Google Scholar] [CrossRef]
- Bertram, S.; Glowacka, I.; Steffen, I.; Kühl, A.; Pöhlmann, S. Novel insights into proteolytic cleavage of influenza virus hemagglutinin. Rev. Med. Virol. 2010, 20, 298–310. [Google Scholar] [CrossRef]
- Böttcher-Friebertshäuser, E.; Freuer, C.; Sielaff, F.; Schmidt, S.; Eickmann, M.; Uhlendorff, J.; Steinmetzer, T.; Klenk, H.D.; Garten, W. Cleavage of Influenza Virus Hemagglutinin by Airway Proteases TMPRSS2 and HAT Differs in Subcellular Localization and Susceptibility to Protease Inhibitors. J. Virol. 2010, 84, 5605–5614. [Google Scholar] [CrossRef] [Green Version]
- Skehel, J.J.; Wiley, D.C. Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annu. Rev. Biochem. 2000, 69, 531–569. [Google Scholar] [CrossRef]
- Varečková, E.; Mucha, V.; Wharton, S.A.; Kostolanský, F. Inhibition of fusion activity of influenza A haemagglutinin mediated by HA2-specific monoclonal antibodies. Arch. Virol. 2003, 148, 469–486. [Google Scholar] [CrossRef]
- Jakubcová, L.; Hollý, J.; Varečková, E. The role of fusion activity of influenza A viruses in their biological properties. Acta Virol. 2016, 60, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Jakubcová, L.; Vozárová, M.; Hollý, J.; Tomčíková, K.; Fogelová, M.; Polčicová, K.; Kostolanský, F.; Fodor, E.; Varečková, E. Biological properties of influenza A virus mutants with amino acid substitutions in the HA2 glycoprotein of the HA1/HA2 interaction region. J. Gen. Virol. 2019, 100, 1282–1292. [Google Scholar] [CrossRef]
- Böttcher-Friebertshäuser, E.; Klenk, H.D.; Garten, W. Activation of influenza viruses by proteases from host cells and bacteria in the human airway epithelium. Pathog. Dis. 2013, 69, 87–100. [Google Scholar] [CrossRef] [Green Version]
- Tashiro, M.; Ciborowski, P.; Klenk, H.D.; Pulverer, G.; Rott, R. Role of Staphylococcus protease in the development of influenza pneumonia. Nature 1987, 325, 536–537. [Google Scholar] [CrossRef]
- Scheiblauer, H.; Reinacher, M.; Tashiro, M.; Rott, R. Interactions between Bacteria and Influenza A Virus in the Development of Influenza Pneumonia. J. Infect. Dis. 1992, 166, 783–791. [Google Scholar] [CrossRef]
- Klenk, H.; Rott, R.; Orlich, M. Further Studies on the Activation of Influenza Virus by Proteolytic Cleavage of the Haemagglutinin. J. Gen. Virol. 1977, 36, 151–161. [Google Scholar] [CrossRef]
- Mancini, D.A.; Mendonça, R.M.; Dias, A.L.; Mendonça, R.Z.; Pinto, J.R. Co-infection between influenza virus and flagellated bacteria. Rev. Inst. Med. Trop. Sao Paulo 2005, 47, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Callan, R.J.; Hartmann, F.A.; West, S.E.; Hinshaw, V.S. Cleavage of influenza A virus H1 hemagglutinin by swine respiratory bacterial proteases. J. Virol. 1997, 71, 7579–7585. [Google Scholar] [CrossRef] [Green Version]
- Berri, F.; Rimmelzwaan, G.F.; Hanss, M.; Albina, E.; Foucault-Grunenwald, M.-L.; Lê, V.B.; Vogelzang-van Trierum, S.E.; Gil, P.; Camerer, E.; Martinez, D.; et al. Plasminogen Controls Inflammation and Pathogenesis of Influenza Virus Infections via Fibrinolysis. PLoS Pathog. 2013, 9, e1003229. [Google Scholar] [CrossRef] [Green Version]
- Tse, L.V.; Marcano, V.C.; Huang, W.; Pocwierz, M.S.; Whittaker, G.R. Plasmin-Mediated Activation of Pandemic H1N1 Influenza Virus Hemagglutinin Is Independent of the Viral Neuraminidase. J. Virol. 2013, 87, 5161–5169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tse, L.V.; Whittaker, G.R. Modification of the hemagglutinin cleavage site allows indirect activation of avian influenza virus H9N2 by bacterial staphylokinase. Virology 2015, 482, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberhard, T.; Kronvall, G.; Ullberg, M. Surface bound plasmin promotes migration of Streptococcus pneumoniae through reconstituted basement membranes. Microb. Pathog. 1999, 26, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Fulde, M.; Steinert, M.; Bergmann, S. Interaction of streptococcal plasminogen binding proteins with the host fibrinolytic system. Front. Cell. Infect. Microbiol. 2013, 3, 85. [Google Scholar] [CrossRef] [Green Version]
- Fulde, M.; Bernardo-García, N.; Rohde, M.; Nachtigall, N.; Frank, R.; Preissner, K.; Klett, J.; Morreale, A.; Chhatwal, G.S.; Hermoso, J.; et al. Pneumococcal phosphoglycerate kinase interacts with plasminogen and its tissue activator. J. Thromb. Haemost. 2014, 111, 401–416. [Google Scholar] [CrossRef] [Green Version]
- Short, K.R.; Habets, M.N.; Hermans, P.W.; Diavatopoulos, D.A. Interactions between Streptococcus pneumoniae and influenza virus: A mutually beneficial relationship? Future Microbiol. 2012, 7, 609–624. [Google Scholar] [CrossRef]
- Passariello, C.; Nencioni, L.; Sgarbanti, R.; Ranieri, D.; Torrisi, M.R.; Ripa, S.; Garaci, E.; Palamara, A.T. Viral hemagglutinin is involved in promoting the internalisation of Staphylococcus aureus into human pneumocytes during influenza A H1N1 virus infection. Int. J. Med. Microbiol. 2011, 301, 97–104. [Google Scholar] [CrossRef]
- Okamoto, S.; Kawabata, S.; Nakagawa, I.; Okuno, Y.; Goto, T.; Sano, K.; Hamada, S. Influenza A Virus-Infected Hosts Boost an Invasive Type of Streptococcus pyogenes Infection in Mice. J. Virol. 2003, 77, 4104–4112. [Google Scholar] [CrossRef] [Green Version]
- Hosaka, Y.; Ikeura, A.; Harada, Y.; Kuroda, K.; Hamayasu, H.; Suzuki, T.; Yamada, K.; Kawase, Y.; Suzuki, Y. Binding of influenza type A viruses to group B Streptococcus and haemagglutination by virus-bound bacteria. J. Electron. Microsc. 2000, 49, 765–773. [Google Scholar] [CrossRef]
- Short, K.R.; Habets, M.N.; Payne, J.; Reading, P.C.; Diavatopoulos, D.A.; Wijburg, O.L. Influenza A virus induced bacterial otitis media is independent of virus tropism for α2,6-linked sialic acid. Virol. J. 2013, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- McAuley, J.L.; Gilbertson, B.P.; Trifkovic, S.; Brown, L.E.; McKimm-Breschkin, J.L. Influenza Virus Neuraminidase Structure and Functions. Front. Microbiol. 2019, 10, 39. [Google Scholar] [CrossRef] [Green Version]
- Castrucci, M.R.; Kawaoka, Y. Biologic importance of neuraminidase stalk length in influenza A virus. J. Virol. 1993, 67, 759–764. [Google Scholar] [CrossRef] [Green Version]
- Kosik, I.; Yewdell, J.W. Influenza Hemagglutinin and Neuraminidase: Yin-Yang Proteins Coevolving to Thwart Immunity. Viruses 2019, 11, 346. [Google Scholar] [CrossRef] [Green Version]
- Barman, S.; Adhikary, L.; Chakrabarti, A.K.; Bernas, C.; Kawaoka, Y.; Nayak, D.P. Role of transmembrane domain and cytoplasmic tail amino acid sequences of influenza a virus neuraminidase in raft association and virus budding. J. Virol. 2004, 78, 5258–5269. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Leser, G.P.; Zhang, J.; Lamb, R.A. Influenza virus hemagglutinin and neuraminidase cytoplasmic tails control particle shape. EMBO J. 1997, 16, 1236–1247. [Google Scholar] [CrossRef] [Green Version]
- Mitnaul, L.J.; Castrucci, M.R.; Murti, K.G.; Kawaoka, Y. The cytoplasmic tail of influenza A virus neuraminidase (NA) affects NA incorporation into virions, virion morphology, and virulence in mice but is not essential for virus replication. J. Virol. 1996, 70, 873–879. [Google Scholar] [CrossRef] [Green Version]
- Wen, F.; Wan, X.F. Influenza Neuraminidase: Underrated Role in Receptor Binding. Trends. Microbiol. 2019, 27, 477–479. [Google Scholar] [CrossRef]
- Du, R.; Cui, Q.; Rong, L. Competitive Cooperation of Hemagglutinin and Neuraminidase during Influenza A Virus Entry. Viruses 2019, 11, 458. [Google Scholar] [CrossRef] [Green Version]
- Su, B.; Wurtzer, S.; Rameix-Welti, M.A.; Dwyer, D.; van der Werf, S.; Naffakh, N.; Clavel, F.; Labrosse, B. Enhancement of the influenza A hemagglutinin (HA)-mediated cell-cell fusion and virus entry by the viral neuraminidase (NA). PLoS ONE 2009, 4, e8495. [Google Scholar] [CrossRef] [Green Version]
- McCombs, J.E.; Kohler, J.J. Pneumococcal Neuraminidase Substrates Identified through Comparative Proteomics Enabled by Chemoselective Labeling. Bioconjug. Chem. 2016, 27, 1013–1022. [Google Scholar] [CrossRef] [Green Version]
- Pettigrew, M.M.; Fennie, K.P.; York, M.P.; Daniels, J.; Ghaffar, F. Variation in the presence of neuraminidase genes among Streptococcus pneumoniae isolates with identical sequence types. Infect. Immun. 2006, 74, 3360–3365. [Google Scholar] [CrossRef] [Green Version]
- Gut, H.; Xu, G.; Taylor, G.L.; Walsh, M.A. Structural basis for Streptococcus pneumoniae NanA inhibition by influenza antivirals zanamivir and oseltamivir carboxylate. J. Mol. Biol. 2011, 409, 496–503. [Google Scholar] [CrossRef]
- Walther, E.; Xu, Z.; Richter, M.; Kirchmair, J.; Grienke, U.; Rollinger, J.M.; Krumbholz, A.; Saluz, H.P.; Pfister, W.; Sauerbrei, A.; et al. Dual Acting Neuraminidase Inhibitors Open New Opportunities to Disrupt the Lethal Synergism between Streptococcus pneumoniae and Influenza Virus. Front. Microbiol. 2016, 7, 357. [Google Scholar] [CrossRef]
- Gubareva, L.V.; Kaiser, L.; Hayden, F.G. Influenza virus neuraminidase inhibitors. Lancet 2000, 355, 827–835. [Google Scholar] [CrossRef]
- Peltola, V.T.; Murti, K.G.; McCullers, J.A. influenza virus neuraminidase contributes to secondary bacterial pneumonia. J. Infect. Dis. 2005, 192, 249–257. [Google Scholar] [CrossRef]
- Nieminen, J.; St-Pierre, C.; Bhaumik, P.; Poirier, F.; Sato, S. Role of galectin-3 in leukocyte recruitment in a murine model of lung infection by Streptococcus pneumoniae. J. Immunol. 2008, 180, 2466–2473. [Google Scholar] [CrossRef] [Green Version]
- Rabinovich, G.A.; Toscano, M.A.; Jackson, S.S.; Vasta, G.R. Functions of cell surface galectin-glycoprotein lattices. Curr. Opin. Struct. Biol. 2007, 17, 513–520. [Google Scholar] [CrossRef] [Green Version]
- Stowell, S.R.; Karmakar, S.; Arthur, C.M.; Ju, T.; Rodrigues, L.C.; Riul, T.B.; Dias-Baruffi, M.; Miner, J.; McEver, R.P.; Cummings, R.D. Galectin-1 induces reversible phosphatidylserine exposure at the plasma membrane. Mol. Biol. Cell. 2009, 20, 1408–1418. [Google Scholar] [CrossRef] [Green Version]
- Vasta, G.R. Roles of galectins in infection. Nat. Rev. Microbiol. 2009, 7, 424–438. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.Y.; Rabinovich, G.A.; Liu, F.T. Galectins: Structure, function and therapeutic potential. Expert Rev. Mol. Med. 2008, 10, e17. [Google Scholar] [CrossRef]
- Yang, Z.S.; Lin, C.Y.; Huang, S.W.; Wang, W.H.; Urbina, A.N.; Tseng, S.P.; Lu, P.L.; Chen, Y.H.; Wang, S.F. Regulatory roles of galectins on influenza A virus and their potential as a therapeutic strategy. Biomed. Pharmacother. 2021, 139, 111713. [Google Scholar] [CrossRef]
- Bao, J.; Wang, X.; Liu, S.; Zou, Q.; Zheng, S.; Yu, F.; Chen, Y. Galectin-1 ameliorates influenza A H1N1pdm09 virus-induced acute lung injury. Front. Microbiol. 2020, 11, 1293. [Google Scholar] [CrossRef]
- Hattori, T.A.T.; Fujioka, Y.; Maruyama, J.; Nakayama, Y.; Ohba, Y.; Niki, T.; Miyazaki, T.; Hirashima, M.; Kida, H. Inhibition of influenza A virus infection by Galectin-9. Jpn. J. Vet. Res. 2013, 61, 5–18. [Google Scholar] [CrossRef]
- Katoh, S.; Ikeda, M.; Shimizu, H.; Mouri, K.; Obase, Y.; Kobashi, Y.; Fukushima, K.; Hirashima, M.; Oka, M. Increased levels of plasma galectin-9 in patients with influenza virus infection. Tohoku J. Exp. Med. 2014, 232, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Sundararajan, A.; Suryawanshi, A.; Kumar, N.; Veiga-Parga, T.; Kuchroo, V.K.; Thomas, P.G.; Sangster, M.Y.; Rouse, B.T. T cell immunoglobulin and mucin protein-3 (Tim-3)/galectin-9 interaction regulates influenza A virus-specific humoral and CD8 T-cell responses. Proc. Natl. Acad. Sci. USA 2011, 108, 19001–19006. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.J.; Wang, S.F.; Weng, I.C.; Hong, M.H.; Lo, T.H.; Jan, J.T.; Hsu, L.C.; Chen, H.Y.; Liu, F.T. Galectin-3 enhances avian H5N1 influenza A virus-induced pulmonary inflammation by promoting NLRP3 inflammasome activation. Am. J. Pathol. 2018, 188, 1031–1042. [Google Scholar] [CrossRef] [Green Version]
- Li, S.W.; Yang, T.C.; Lai, C.C.; Huang, S.H.; Liao, J.M.; Wan, L.; Lin, Y.J.; Lin, C.W. Antiviral activity of aloe-emodin against influenza A virus via galectin-3 up-regulation. Eur. J. Pharmacol. 2014, 738, 125–132. [Google Scholar] [CrossRef]
- Lin, C.Y.; Yang, Z.S.; Wang, W.H.; Urbina, A.N.; Lin, Y.T.; Huang, J.C.; Liu, F.T.; Wang, S.F. The Antiviral Role of Galectins toward Influenza A Virus Infection—An Alternative Strategy for Influenza Therapy. Pharmaceuticals 2021, 14, 490. [Google Scholar] [CrossRef]
- Wang, H.; Huang, W.; Orwenyo, J.; Banerjee, A.; Vasta, G.R.; Wang, L.X. Design and synthesis of glycoprotein-based multivalent glyco-ligands for influenza hemagglutinin and human galectin-3. Bioorg. Med. Chem. 2013, 21, 2037–2044. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.H.; Lin, C.Y.; Chang, M.R.; Urbina, A.N.; Assavalapsakul, W.; Thitithanyanont, A.; Chen, Y.H.; Liu, F.T.; Wang, S.F. The role of galectins in virus infection—A systemic literature review. J. Microbiol. Immunol. Infect. 2020, 53, 925–935. [Google Scholar] [CrossRef]
- Brydon, E.W.; Morris, S.J.; Sweet, C. Role of apoptosis and cytokines in influenza virus morbidity. FEMS Microbiol. Rev. 2005, 29, 837–850. [Google Scholar] [CrossRef] [Green Version]
- Schultz-Cherry, S.; Hinshaw, V.S. Influenza virus neuraminidase activates latent transforming growth factor beta. J. Virol. 1996, 70, 8624–8629. [Google Scholar] [CrossRef] [Green Version]
- Carlson, C.M.; Turpin, E.A.; Moser, L.A.; O’Brien, K.B.; Cline, T.D.; Jones, J.C.; Tumpey, T.M.; Katz, J.M.; Kelley, L.A.; Gauldie, J.; et al. Transforming growth factor-β: Activation by neuraminidase and role in highly pathogenic H5N1 influenza pathogenesis. PLoS Pathog. 2010, 6, e1001136. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Huang, S.; Chen, J.; Zhang, S.; Chen, Z. NA proteins of influenza A viruses H1N1/2009, H5N1, and H9N2 show differential effects on infection initiation, virus release, and cell-cell fusion. PLoS ONE 2013, 8, e54334. [Google Scholar] [CrossRef] [Green Version]
- Letterio, J.J.; Roberts, A.B. Regulation of immune responses by TGF-beta. Annu. Rev. Immunol. 1998, 16, 137–161. [Google Scholar] [CrossRef] [Green Version]
- Roberts, S.; Salmon, S.L.; Steiner, D.J.; Williams, C.M.; Metzger, D.W.; Furuya, Y. Allergic Airway Disease Prevents Lethal Synergy of Influenza A Virus-Streptococcus pneumoniae Coinfection. MBio 2019, 10, e01335-19. [Google Scholar] [CrossRef] [Green Version]
- Hsu, A.C. Influenza Virus: A Master Tactician in Innate Immune Evasion and Novel Therapeutic Interventions. Front. Immunol. 2018, 9, 743. [Google Scholar] [CrossRef] [Green Version]
- Jia, D.; Rahbar, R.; Chan, R.W.; Lee, S.M.; Chan, M.C.; Wang, B.X.; Baker, D.P.; Sun, B.; Peiris, J.S.; Nicholls, J.M.; et al. Influenza virus non-structural protein 1 (NS1) disrupts interferon signaling. PLoS ONE 2010, 5, e13927. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Jeong, M.S.; Jang, S.B. Structure and Activities of the NS1 Influenza Protein and Progress in the Development of Small-Molecule Drugs. Int. J. Mol. Sci. 2021, 22, 4242. [Google Scholar] [CrossRef]
- Ayllon, J.; García-Sastre, A. The NS1 protein: A multitasking virulence factor. Curr. Top. Microbiol. Immunol. 2015, 386, 73–107. [Google Scholar] [CrossRef]
- Krug, R.M. Functions of the influenza A virus NS1 protein in antiviral defense. Curr. Opin. Virol. 2015, 12, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Sesma, A. The influenza virus NS1 protein: Inhibitor of innate and adaptive immunity. Infect. Disord. Drug Targets 2007, 7, 336–343. [Google Scholar] [CrossRef]
- Fernandez-Sesma, A.; Marukian, S.; Ebersole, B.J.; Kaminski, D.; Park, M.S.; Yuen, T.; Sealfon, S.C.; García-Sastre, A.; Moran, T.M. Influenza virus evades innate and adaptive immunity via the NS1 protein. J. Virol. 2006, 80, 6295–6304. [Google Scholar] [CrossRef] [Green Version]
- García-Sastre, A.; Egorov, A.; Matassov, D.; Brandt, S.; Levy, D.E.; Durbin, J.E.; Palese, P.; Muster, T. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 1998, 252, 324–330. [Google Scholar] [CrossRef] [Green Version]
- Shepardson, K.; Larson, K.; Cho, H.; Johns, L.L.; Malkoc, Z.; Stanek, K.; Wellhman, J.; Zaiser, S.; Daggs-Olson, J.; Moodie, T.; et al. A Novel Role for PDZ-Binding Motif of Influenza A Virus Nonstructural Protein 1 in Regulation of Host Susceptibility to Postinfluenza Bacterial Superinfections. Viral Immunol. 2019, 32, 131–143. [Google Scholar] [CrossRef]
- Hu, M.M.; Shu, H.B. Cytoplasmic Mechanisms of Recognition and Defense of Microbial Nucleic Acids. Annu. Rev. Cell Dev. Biol. 2018, 34, 357–379. [Google Scholar] [CrossRef]
- Sun, X.; Feng, W.; Guo, Y.; Wang, Q.; Dong, C.; Zhang, M.; Guan, Z.; Duan, M. MCPIP1 attenuates the innate immune response to influenza A virus by suppressing RIG-I expression in lung epithelial cells. J. Med. Virol. 2018, 90, 204–211. [Google Scholar] [CrossRef]
- Guo, Z.; Chen, L.M.; Zeng, H.; Gomez, J.A.; Plowden, J.; Fujita, T.; Katz, J.M.; Donis, R.O.; Sambhara, S. NS1 protein of influenza A virus inhibits the function of intracytoplasmic pathogen sensor, RIG-I. Am. J. Respir. Cell Mol. 2007, 36, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Mibayashi, M.; Martínez-Sobrido, L.; Loo, Y.M.; Cárdenas, W.B.; Gale, M., Jr.; García-Sastre, A. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J. Virol. 2007, 81, 514–524. [Google Scholar] [CrossRef] [Green Version]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Näslund, T.I.; Liljeström, P.; Weber, F.; Reis e Sousa, C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef] [Green Version]
- Weber-Gerlach, M.; Weber, F. To conquer the host, influenza virus is packing it in: Interferon-antagonistic strategies beyond NS1. J. Virol. 2016, 90, 8389–8394. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, S.; Planz, O. Influenza viruses and the NF-kappaB signaling pathway—towards a novel concept of antiviral therapy. Biol. Chem. 2008, 389, 1307–1312. [Google Scholar] [CrossRef]
- DiDonato, J.A.; Hayakawa, M.; Rothwarf, D.M.; Zandi, E.; Karin, M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature 1997, 388, 548–554. [Google Scholar] [CrossRef]
- Zamanian-Daryoush, M.; Mogensen, T.H.; DiDonato, J.A.; Williams, B.R. NF-kappaB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-kappaB-inducing kinase and IkappaB kinase. Mol. Cell. Biol. 2000, 20, 1278–1290. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, M.; Zheng, H.; Muster, T.; Palese, P.; Beg, A.A.; García-Sastre, A. Influenza A virus NS1 protein prevents activation of NF-kappaB and induction of alpha/beta interferon. J. Virol. 2000, 74, 11566–11573. [Google Scholar] [CrossRef] [Green Version]
- Rynda-Apple, A.; Robinson, K.M.; Alcorn, J.F. Influenza and Bacterial Superinfection: Illuminating the Immunologic Mechanisms of Disease. Infect Immun. 2015, 83, 3764–3770. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
| IAV Protein | Function | Functional Domain | References |
|---|---|---|---|
| PB1-F2 | Inhibition of IFN response | N66S mutation | [148,149] |
| Apoptosis of epithelial and immune cells | - | [150,151] | |
| Cytotoxic death of epithelial and immune cells | Cytotoxic motif | [152] | |
| Induction of rapid inflammatory response | Inflammatory motif | [153,154] | |
| Regulation of NLRP3 inflammasome activity | - | [155,156,157,158] | |
| HA | Regulation of autophagosome formation | - | [119,127,159,160] |
| NA | Creation of environment for bacterial entrance | ||
| Alteration of glycosylation on cell surface | Catalytic domain | [138,139] | |
| Desialylation of surface glycans | [161,162] | ||
| Affection of innate immunity | |||
| Direct activation of TGF-β | Catalytic domain | [163,164,165] | |
| NS1 | Inhibition of IFN response in several ways | ||
| Blocking of RIG-I activation | RNA-binding domain | [166,167,168] | |
| Blocking of PKR activation | [168,169,170] | ||
| Blocking OAS function | [171,172] | ||
| Interaction with host factors | Effector domain PDZ-binding motif | [173,174,175,176] | |
| Manipulation of apoptosis in several ways | |||
| Binding to PKR linker domain | Effector domain | [120,132] | |
| Activation of PI3K pathway | SH3-binding motif aa 164–167 | [119,177,178,179] | |
| Interaction with Hsp90 | - | [180,181] | |
| Inhibition of p53 | aa 144–188 | [182,183,184] | |
| M2 | Induction of autophagosome formation | - | [119,124,129] |
| Inhibition of lysosomal degradation of autophagosomes | - | [129,185,186] | |
| NP | Induction of autophagosome formation | - | [128,129] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikušová, M.; Tomčíková, K.; Briestenská, K.; Kostolanský, F.; Varečková, E. The Contribution of Viral Proteins to the Synergy of Influenza and Bacterial Co-Infection. Viruses 2022, 14, 1064. https://doi.org/10.3390/v14051064
Mikušová M, Tomčíková K, Briestenská K, Kostolanský F, Varečková E. The Contribution of Viral Proteins to the Synergy of Influenza and Bacterial Co-Infection. Viruses. 2022; 14(5):1064. https://doi.org/10.3390/v14051064
Chicago/Turabian StyleMikušová, Miriam, Karolína Tomčíková, Katarína Briestenská, František Kostolanský, and Eva Varečková. 2022. "The Contribution of Viral Proteins to the Synergy of Influenza and Bacterial Co-Infection" Viruses 14, no. 5: 1064. https://doi.org/10.3390/v14051064
APA StyleMikušová, M., Tomčíková, K., Briestenská, K., Kostolanský, F., & Varečková, E. (2022). The Contribution of Viral Proteins to the Synergy of Influenza and Bacterial Co-Infection. Viruses, 14(5), 1064. https://doi.org/10.3390/v14051064