
Convolutional Neural Networks Based on Sequential Spike Predict the High Human Adaptation of SARS-CoV-2 Omicron Variants

 , and
, and
Abstract
:1. Introduction
2. Methods
2.1. Preparation of Spike Sequences and Representation of Sequential Amino Acid (AA)
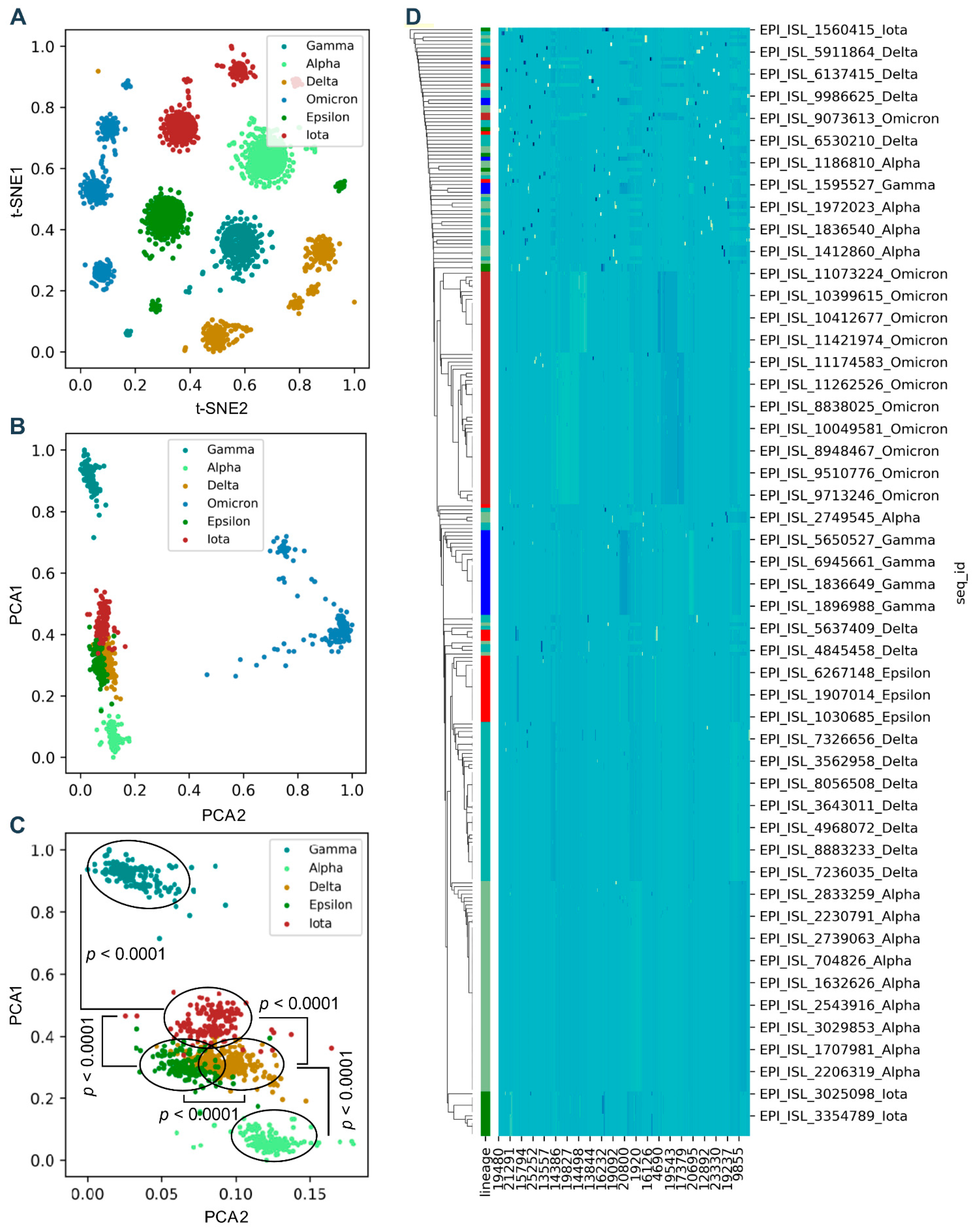
2.2. Unsupervised Machine Learning (ML)
2.3. Model Architecture of a 2D-CNN
2.4. Supplementary Data and Code Availability
2.5. Data Analysis or Statistical Analysis
3. Results
3.1. Methodology of a Deep Learning Model to Predict Human Adaptation of SARS-CoV-2 Variants
3.2. Genomic Codons Discriminate the Difference between SARS-CoV-2 Variants
3.3. Importance of Human Adaptation-Associated Codons in the Spike of SARS-CoV-2
3.4. Performance of the Deep Learning Model to Predict SARS-CoV-2 Adaptation
3.5. Human Adaptation of SARS-CoV-2 Omicron Variants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Abbreviations | Full Names |
| CNN | Convolutional Neural Network |
| COVID-19 | coronavirus disease 2019 |
| SARS-CoV-2 | severe acute respiratory coronavirus 2 |
| VOC | variant of concern |
| R0 | basic reproductive number |
| ML | machine learning |
| DL | deep learning |
| DNT | dinucleotide |
| DCR | DNT representation |
| AA | amino acid |
| t-SNE | t-distributed stochastic neighbor embedding |
| PCA | Principal Component Analysis |
References
- WHO Coronavirus (COVID-19) Dashboard. 2022. Available online: https://covid19.who.int/ (accessed on 28 March 2022).
- Chaqroun, A.; Hartard, C.; Schvoerer, E. Anti-SARS-CoV-2 Vaccines and Monoclonal Antibodies Facing Viral Variants. Viruses 2021, 13, 1171. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef] [PubMed]
- Jansen, L.; Tegomoh, B.; Lange, K.; Showalter, K.; Figliomeni, J.; Abdalhamid, B.; Iwen, P.C.; Fauver, J.; Buss, B.; Donahue, M. Investigation of a SARS-CoV-2 B.1.1.529 (Omicron) Variant Cluster—Nebraska, November–December 2021. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 1782–1784. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Rocklov, J. The effective reproduction number for the omicron SARS-CoV-2 variant of concern is several times higher than Delta. J. Travel Med. 2022, taac037. [Google Scholar] [CrossRef]
- Liu, Y.; Rocklöv, J. The reproductive number of the Delta variant of SARS-CoV-2 is far higher compared to the ancestral SARS-CoV-2 virus. J. Travel Med. 2021, 28, taab124. [Google Scholar] [CrossRef]
- Sigal, A.; Milo, R.; Jassat, W. Estimating disease severity of Omicron and Delta SARS-CoV-2 infections. Nat. Rev. Immunol. 2022, 22, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Menni, C.; Valdes, A.M.; Polidori, L.; Antonelli, M.; Penamakuri, S.; Nogal, A.; Louca, P.; May, A.; Figueiredo, J.C.; Hu, C.; et al. Symptom prevalence, duration, and risk of hospital admission in individuals infected with SARS-CoV-2 during periods of omicron and delta variant dominance: A prospective observational study from the ZOE COVID Study. Lancet 2022, 399, 1618–1624. [Google Scholar] [CrossRef]
- He, C.; He, X.; Yang, J.; Lei, H.; Hong, W.; Song, X.; Yang, L.; Li, J.; Wang, W.; Shen, G.; et al. Spike protein of SARS-CoV-2 Omicron (B.1.1.529) variant have a reduced ability to induce the immune response. Signal Transduct. Target. Ther. 2022, 7, 119. [Google Scholar] [CrossRef]
- Sheward, D.J.; Kim, C.; Ehling, R.A.; Pankow, A.; Castro, D.X.; Dyrdak, R.; Martin, D.P.; Reddy, S.T.; Dillner, J.; Karlsson, H.G.; et al. Neutralisation sensitivity of the SARS-CoV-2 omicron (B.1.1.529) variant: A cross-sectional study. Lancet Infect. Dis. 2022. [Google Scholar] [CrossRef]
- Ortega, J.T.; Jastrzebska, B.; Rangel, H.R. Omicron SARS-CoV-2 Variant Spike Protein Shows an Increased Affinity to the Human ACE2 Receptor: An In Silico Analysis. Pathogens 2022, 11, 45. [Google Scholar] [CrossRef]
- Vardhan, S.; Sahoo, S.K. Computational studies on the interaction of SARS-CoV-2 Omicron SGp RBD with human receptor ACE2, limonin and glycyrrhizic acid. Comput. Biol. Med. 2022, 144, 105367. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D. Will Omicron finally overpower China’s COVID defences? Nature 2022, 604, 17–18. [Google Scholar] [CrossRef] [PubMed]
- Ozono, S.; Zhang, Y.; Ode, H.; Sano, K.; Tan, T.S.; Imai, K.; Miyoshi, K.; Kishigami, S.; Ueno, T.; Iwatani, Y.; et al. SARS-CoV-2 D614G spike mutation increases entry efficiency with enhanced ACE2-binding affinity. Nat. Commun. 2021, 12, 848. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhao, T.; Lu, M.; Zhang, Y.; Yao, X.; Wu, G.; Dai, F.; Zhang, F.; Zhang, G. The Analyses of High Infectivity Mechanism of SARS-CoV-2 and Its Variants. COVID 2021, 1, 666–673. [Google Scholar] [CrossRef]
- Cele, S.; Gazy, I.; Jackson, L.; Hwa, S.H.; Tegally, H.; Lustig, G.; Giandhari, J.; Pillay, S.; Wilkinson, E.; Naidoo, Y.; et al. Escape of SARS-CoV-2 501Y.V2 from neutralization by convalescent plasma. Nature 2021, 593, 142–146. [Google Scholar] [CrossRef]
- Torbati, E.; Krause, K.L.; Ussher, J.E. The Immune Response to SARS-CoV-2 and Variants of Concern. Viruses 2021, 13, 1911. [Google Scholar] [CrossRef]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef]
- Li, J.; Zhang, S.; Li, B.; Hu, Y.; Kang, X.P.; Wu, X.Y.; Huang, M.T.; Li, Y.C.; Zhao, Z.P.; Qin, C.F.; et al. Machine Learning Methods for Predicting Human-Adaptive Influenza A Viruses Based on Viral Nucleotide Compositions. Mol. Biol. Evol. 2020, 37, 1224–1236. [Google Scholar] [CrossRef]
- Taubenberger, J.K.; Kash, J.C. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe 2010, 7, 440–451. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wu, Y.N.; Zhang, S.; Kang, X.P.; Jiang, T. Deep learning based on biologically interpretable genome representation predicts two types of human adaptation of SARS-CoV-2 variants. Brief. Bioinform. 2022, bbac036. [Google Scholar] [CrossRef]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babayan, S.A.; Orton, R.J.; Streicker, D.G. Predicting reservoir hosts and arthropod vectors from evolutionary signatures in RNA virus genomes. Science 2018, 362, 577–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, X. Extreme Genomic CpG Deficiency in SARS-CoV-2 and Evasion of Host Antiviral Defense. Mol. Biol. Evol. 2020, 37, 2699–2705. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- Jolliffe, I.T.; Cadima, J. Principal component analysis: A review and recent developments. Philos. Trans. A Math. Phys. Eng. Sci. 2016, 374, 20150202. [Google Scholar] [CrossRef]
- Metz, C.E. Basic principles of ROC analysis. Semin. Nucl. Med. 1978, 8, 283–298. [Google Scholar] [CrossRef]
- Bahir, I.; Fromer, M.; Prat, Y.; Linial, M. Viral adaptation to host: A proteome-based analysis of codon usage and amino acid preferences. Mol. Syst. Biol. 2009, 5, 311. [Google Scholar] [CrossRef]
- Manz, B.; Schwemmle, M.; Brunotte, L. Adaptation of avian influenza A virus polymerase in mammals to overcome the host species barrier. J. Virol. 2013, 87, 7200–7209. [Google Scholar] [CrossRef] [Green Version]
- Mahase, E. Omicron sub-lineage BA.2 may have “substantial growth advantage,” UKHSA reports. BMJ 2022, 376, o263. [Google Scholar] [CrossRef]
- Hirotsu, Y.; Maejima, M.; Shibusawa, M.; Natori, Y.; Nagakubo, Y.; Hosaka, K.; Sueki, H.; Mochizuki, H.; Tsutsui, T.; Kakizaki, Y.; et al. SARS-CoV-2 Omicron sublineage BA.2 replaces BA.1.1: Genomic surveillance in Japan from September 2021 to March 2022. J. Infect. 2022. [Google Scholar] [CrossRef]
- Chen, J.; Wei, G. Omicron BA.2 (B.1.1.529.2): High Potential for Becoming the Next Dominant Variant. J. Phys. Chem. Lett. 2022, 13, 3840–3849. [Google Scholar] [CrossRef]
- Butt, A.A.; Dargham, S.R.; Loka, S.; Shaik, R.M.; Chemaitelly, H.; Tang, P.; Hasan, M.R.; Coyle, P.V.; Yassine, H.M.; Al-Khatib, H.A.; et al. COVID-19 Disease Severity in Children Infected with the Omicron Variant. Clin. Infect. Dis. 2022, ciac275. [Google Scholar] [CrossRef] [PubMed]
- Arora, P.; Zhang, L.; Rocha, C.; Sidarovich, A.; Kempf, A.; Schulz, S.; Cossmann, A.; Manger, B.; Baier, E.; Tampe, B.; et al. Comparable neutralisation evasion of SARS-CoV-2 omicron subvariants BA.1, BA.2, and BA.3. Lancet Infect. Dis. 2022. [Google Scholar] [CrossRef]
- Anichini, G.; Terrosi, C.; Gori Savellini, G.; Gandolfo, C.; Franchi, F.; Cusi, M.G. Neutralizing Antibody Response of Vaccinees to SARS-CoV-2 Variants. Vaccines 2021, 9, 517. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, C.; Mayer, C.K.; Claassen, M.; Maponga, T.; Burgers, W.A.; Keeton, R.; Riou, C.; Sutherland, A.D.; Suliman, T.; Shaw, M.L.; et al. Breakthrough infections with SARS-CoV-2 omicron despite mRNA vaccine booster dose. Lancet 2022, 399, 625–626. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sublineages | Total | Adaptation 0 | Adaptation 1 | Adaptation 2 |
|---|---|---|---|---|
| BA.1 | 28,412 | 1 | 27,986 | 425 |
| BA.2 | 11,497 | 0 | 208 | 11,289 |
| BA.3 | 10 | 0 | 10 | 0 |
| Nan | 81 | 0 | 37 | 44 |
| Total | 40,000 | 1 | 28,241 | 11,758 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nan, B.-G.; Zhang, S.; Li, Y.-C.; Kang, X.-P.; Chen, Y.-H.; Li, L.; Jiang, T.; Li, J. Convolutional Neural Networks Based on Sequential Spike Predict the High Human Adaptation of SARS-CoV-2 Omicron Variants. Viruses 2022, 14, 1072. https://doi.org/10.3390/v14051072
Nan B-G, Zhang S, Li Y-C, Kang X-P, Chen Y-H, Li L, Jiang T, Li J. Convolutional Neural Networks Based on Sequential Spike Predict the High Human Adaptation of SARS-CoV-2 Omicron Variants. Viruses. 2022; 14(5):1072. https://doi.org/10.3390/v14051072
Chicago/Turabian StyleNan, Bei-Guang, Sen Zhang, Yu-Chang Li, Xiao-Ping Kang, Yue-Hong Chen, Lin Li, Tao Jiang, and Jing Li. 2022. "Convolutional Neural Networks Based on Sequential Spike Predict the High Human Adaptation of SARS-CoV-2 Omicron Variants" Viruses 14, no. 5: 1072. https://doi.org/10.3390/v14051072