
Functional Analysis of a Frontal miRNA Cluster Located in the Large Latency Transcript of Pseudorabies Virus



Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Virus Infection
2.2. Generation of an ST Cell Line for Constitutive PRV miRNA Expression
2.3. Cell Viability Assay
2.4. RNA Extraction and Quantitative Reverse Transcription PCR (RT-qPCR)
2.5. Viral Replication Kinetics
2.6. Protein Extraction and Immunoblotting
2.7. Immunofluorescence Imaging
2.8. Flow Cytometry
2.9. Statistical Data Analysis
3. Results
3.1. Generation and Characterization of the ST Cell Line Constitutively Expressing PRV LLT [1–3] miRNAs
3.2. Expression Profiles of LLT [1–3] miRNAs during PRV Infection
3.3. IE180 and EP0 Expression Is Downregulated by the Overexpression of LLT [1–3] miRNAs
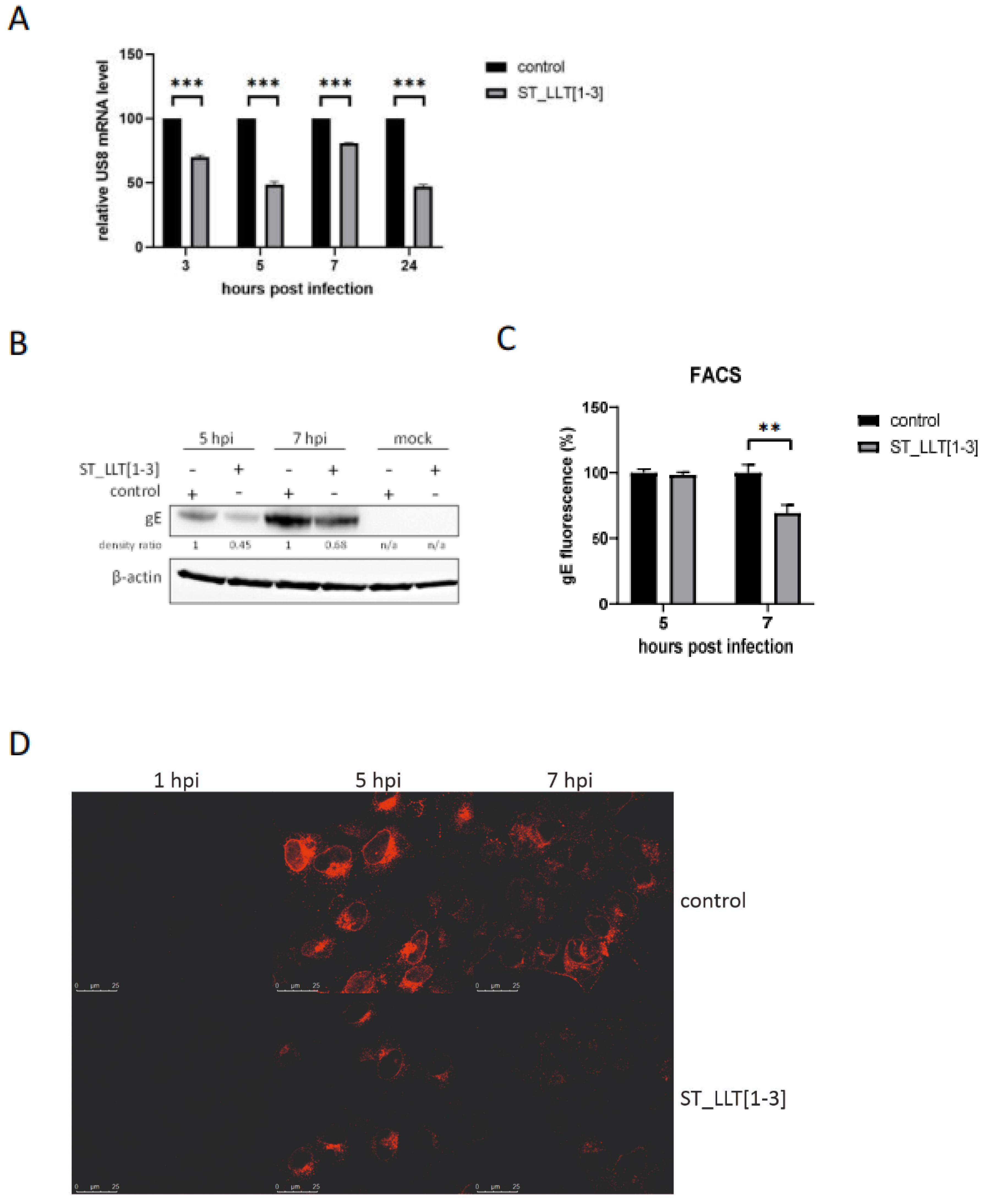
3.4. Production of gE Is Hampered by LLT [1–3] miRNAs
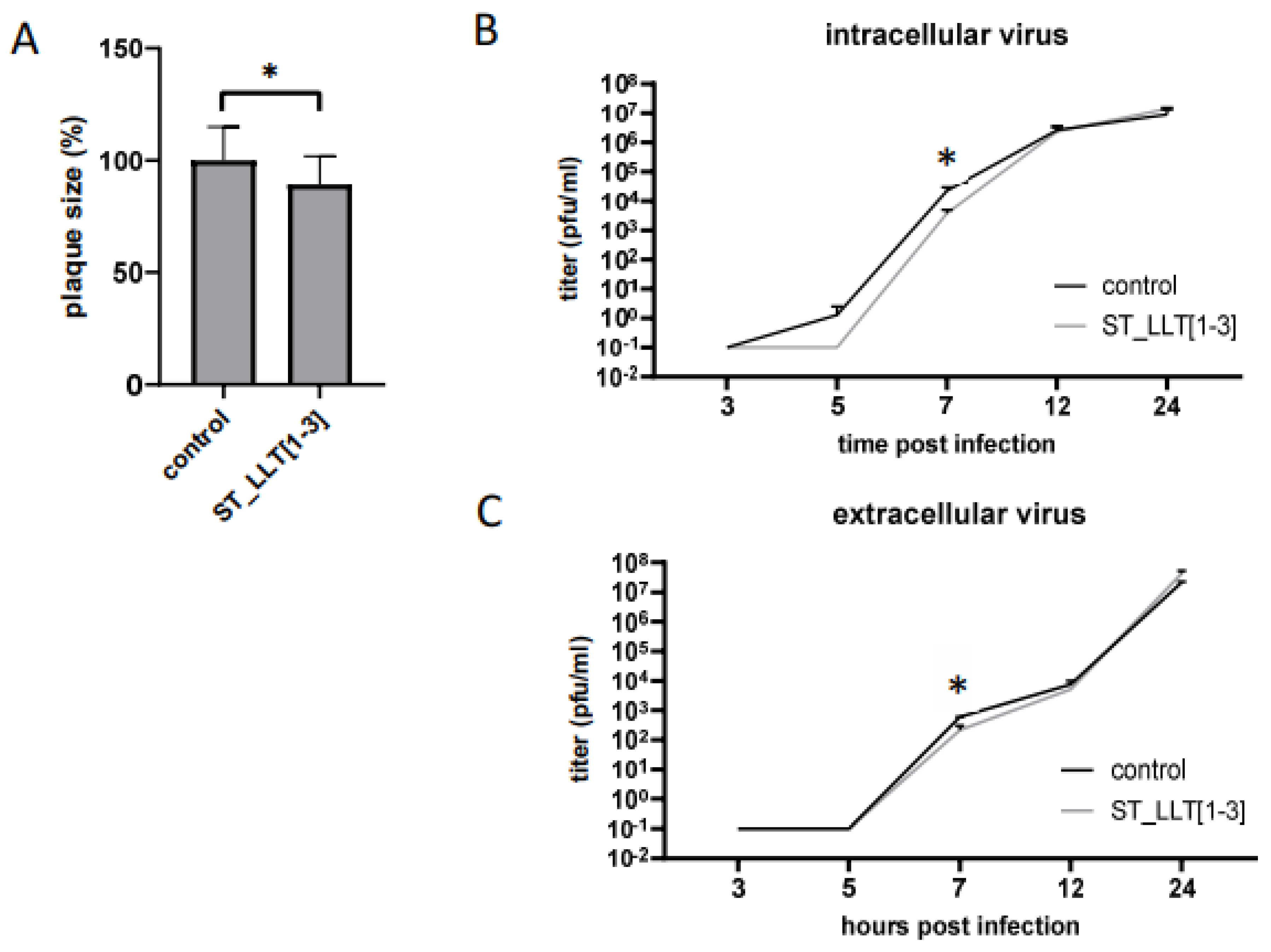
3.5. The Effect of LLT [1–3] miR Cluster Overexpression on PRV Replication
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mettenleiter, T.C. Pseudorabies (Aujeszky’s disease) virus: State of the art. August 1993. Acta Vet. Hung. 1994, 42, 153–177. [Google Scholar] [PubMed]
- Müller, T.; Hahn, E.C.; Tottewitz, F.; Kramer, M.; Klupp, B.G.; Mettenleiter, T.C.; Freuling, C. Pseudorabies virus in wild swine: A global perspective. Arch. Virol. 2011, 156, 1691–1705. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Yao, J.; Yang, Y.; Luo, W.; Yuan, X.; Yang, L.; Wang, A. Current status and challenge of pseudorabies virus infection in china. Virol. Sin. 2021, 36, 588–607. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Guan, H.; Li, C.; Li, Y.; Wang, S.; Zhao, X.; Zhao, Y.; Liu, Y. Characteristics of human encephalitis caused by pseudorabies virus: A case series study. Int. J. Infect. Dis. 2019, 87, 92–99. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Chen, X.X.; Zhang, G. Human PRV Infection in China: An alarm to accelerate eradication of prv in domestic pigs. Virol. Sin. 2021, 36, 823–828. [Google Scholar] [CrossRef]
- Pomeranz, L.E.; Reynolds, A.E.; Hengartner, C.J. Molecular biology of pseudorabies virus: Impact on neurovirology and veterinary medicine. Microbiol. Mol. Biol. Rev. 2005, 69, 462–500. [Google Scholar] [CrossRef] [Green Version]
- Ono, E.; Watanabe, S.; Nikami, H.; Tasaki, T.; Kida, H. Pseudorabies virus (PRV) early protein 0 activates PRV gene transcription in combination with the immediate-early protein IE180 and enhances the infectivity of PRV genomic DNA. Vet. Microbiol. 1998, 63, 99–107. [Google Scholar] [CrossRef]
- Wu, B.W.; Engel, E.A.; Enquist, L.W. Characterization of a replication-incompetent pseudorabies virus mutant lacking the sole immediate early gene IE180. mBio 2014, 5, e01850. [Google Scholar] [CrossRef] [Green Version]
- Moriuchi, H.; Moriuchi, M.; Dean, H.; Cheung, A.K.; Cohen, J.I. Pseudorabies virus EP0 is functionally homologous to varicella-zoster virus ORF61 protein and herpes simplex virus type 1 ICP0. Virology 1995, 209, 281–283. [Google Scholar] [CrossRef] [Green Version]
- Knipe, D.M.; Raja, P.; Lee, J.S. Clues to mechanisms of herpesviral latent infection and potential cures. Proc. Natl. Acad. Sci. USA 2015, 112, 11993–11994. [Google Scholar] [CrossRef] [Green Version]
- Priola, S.A.; Gustafson, D.P.; Wagner, E.K.; Stevens, J.G. A major portion of the latent pseudorabies virus genome is transcribed in trigeminal ganglia of pigs. J. Virol. 1990, 64, 4755–4760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 2008, 454, 780–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, S.; Bertke, A.S.; Patel, A.; Wang, K.; Cohen, J.I.; Krause, P.R. An acutely and latently expressed herpes simplex virus 2 viral microRNA inhibits expression of ICP34.5, a viral neurovirulence factor. Proc. Natl. Acad. Sci. USA 2008, 105, 10931–10936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, S.; Zavolan, M.; Grässer, F.A.; Chien, M.; Russo, J.J.; Ju, J.; John, B.; Enright, A.J.; Mark, D.; Sander, C.; et al. Identification of virus-encoded microRNAs. Science 2004, 304, 734–736. [Google Scholar] [CrossRef]
- Cox, J.E.; Sullivan, C.S. Balance and Stealth: The role of noncoding RNAs in the regulation of virus gene expression. Annu. Rev. Virol. 2014, 1, 89–109. [Google Scholar] [CrossRef] [Green Version]
- Sorel, O.; Dewals, B.G. MicroRNAs in large herpesvirus DNA genomes: Recent advances. Biomol. Concepts 2016, 7, 229–239. [Google Scholar] [CrossRef] [Green Version]
- Mishra, R.; Kumar, A.; Ingle, H.; Kumar, H. The Interplay Between Viral-Derived miRNAs and Host Immunity During Infection. Front. Immunol. 2020, 10, 3079. [Google Scholar] [CrossRef] [Green Version]
- Cheung, A.K. Cloning of the latency gene and the early protein 0 gene of pseudorabies virus. J. Virol. 1991, 65, 5260–5271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.; Scherba, G. Expression of the pseudorabies virus latency-associated transcript gene during productive infection of cultured cells. J. Virol. 1999, 73, 9781–9788. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.Q.; Chen, D.J.; He, H.B.; Chen, D.S.; Chen, L.L.; Chen, H.C.; Liu, Z.F. Pseudorabies virus infected porcine epithelial cell line generates a diverse set of host microRNAs and a special cluster of viral microRNAs. PLoS ONE 2012, 7, e30988. [Google Scholar] [CrossRef] [Green Version]
- Anselmo, A.; Flori, L.; Jaffrezic, F.; Rutigliano, T.; Cecere, M.; Cortes-Perez, N.; Lefèvre, F.; Rogel-Gaillard, C.; Giuffra, E. Co-expression of host and viral microRNAs in porcine dendritic cells infected by the pseudorabies virus. PLoS ONE 2011, 6, e17374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timoneda, O.; Núñez-Hernández, F.; Balcells, I.; Muñoz, M.; Castelló, A.; Vera, G.; Pérez, L.J.; Egea, R.; Mir, G.; Córdoba, S.; et al. The role of viral and host microRNAs in the Aujeszky’s disease virus during the infection process. PLoS ONE 2014, 9, e86965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Zheng, H.; Tong, W.; Li, G.X.; Tian, Q.; Liang, C.; Li, L.W.; Zheng, X.C.; Tong, G.Z. Identification and analysis of novel viral and host dysregulated microRNAs in variant pseudorabies virus-infected PK15 cells. PLoS ONE 2016, 11, e0151546. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zheng, G.; Zhang, Y.; Yang, X.; Liu, H.; Chang, H.; Wang, X.; Zhao, J.; Wang, C.; Chen, L. MicroRNA analysis in mouse neuro-2a cells after pseudorabies virus infection. J. Neurovirol. 2017, 23, 430–440. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, M.M.; Yan, K.; Tang, Q.; Wu, Y.Q.; He, W.B.; Chen, H.C.; Liu, Z.F. The full-length microRNA cluster in the intron of large latency transcript is associated with the virulence of pseudorabies virus. Virology 2018, 520, 59–66. [Google Scholar] [CrossRef]
- Liu, H.; Yang, L.; Shi, Z.; Lv, R.; Yang, X.; Wang, C.; Chen, L.; Chang, H. Functional analysis of prv-miR-LLT11a encoded by pseudorabies virus. J. Vet. Sci. 2019, 20, e68. [Google Scholar] [CrossRef]
- Koppers-Lalic, D.; Reits, E.A.; Ressing, M.E.; Lipinska, A.D.; Abele, R.; Koch, J.; Marcondes Rezende, M.; Admiraal, P.; van Leeuwen, D.; Bienkowska-Szewczyk, K.; et al. Varicelloviruses avoid T cell recognition by UL49.5-mediated inactivation of the transporter associated with antigen processing. Proc. Natl. Acad. Sci. USA 2005, 102, 5144–5149. [Google Scholar] [CrossRef] [Green Version]
- Van Beveren, C.; Goddard, J.G.; Berns, A.; Verma, I.M. Structure of Moloney murine leukemia viral DNA: Nucleotide sequence of the 5’ long terminal repeat and adjacent cellular sequences. Proc. Natl. Acad. Sci. USA 1980, 77, 3307–3311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tombácz, D.; Csabai, Z.; Oláh, P.; Balázs, Z.; Likó, I.; Zsigmond, L.; Sharon, D.; Snyder, M.; Boldogkői, Z. Full-length isoform sequencing reveals novel transcripts and substantial transcriptional overlaps in a herpesvirus. PLoS ONE 2016, 11, e0162868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, Q.; Li, J.; Guo, Y.; Yan, F.; Liu, X.; Blissard, G.W.; Li, Z. Efficient entry of budded virions of Autographa californica multiple nucleopolyhedrovirus into Spodoptera frugiperda cells is dependent on dynamin, Rab5, and Rab11. Insect Biochem. Mol. Biol. 2020, 123, 103409. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Song, H.; Guan, K.; Zhou, J.; Xia, X.; Li, F. Characterization of swine testicular cell line as immature porcine Sertoli cell line. Vitro Cell Dev. Biol. Anim. 2016, 52, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Favoreel, H.W.; Van Minnebruggen, G.; Adriaensen, D.; Nauwynck, H.J. Cytoskeletal rearrangements and cell extensions induced by the US3 kinase of an alphaherpesvirus are associated with enhanced spread. Proc. Natl. Acad. Sci. USA 2005, 102, 8990–8995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Minnebruggen, G.; Favoreel, H.W.; Jacobs, L.; Nauwynck, H.J. Pseudorabies virus US3 protein kinase mediates actin stress fiber breakdown. J. Virol. 2003, 77, 9074–9080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathijs, E.; Vandenbussche, F.; Verpoest, S.; De Regge, N.; Van Borm, S. Complete genome sequence of pseudorabies virus reference strain NIA3 using single-molecule real-time sequencing. Genome Announc. 2016, 4, e00440-16. [Google Scholar] [CrossRef] [Green Version]
- Grimson, A.; Farh, K.K.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol. Cell 2007, 27, 91–105. [Google Scholar] [CrossRef] [Green Version]
- Tombácz, D.; Balázs, Z.; Csabai, Z.; Moldován, N.; Szűcs, A.; Sharon, D.; Snyder, M.; Boldogkői, Z. Characterization of the dynamic transcriptome of a herpesvirus with long-read single molecule real-time sequencing. Sci. Rep. 2017, 7, 43751. [Google Scholar] [CrossRef]
- Wang, C.H.; Yuan, J.; Qin, H.Y.; Luo, Y.; Cong, X.; Li, Y.; Chen, J.; Li, S.; Sun, Y.; Qiu, H.J. A novel gE-deleted pseudorabies virus (PRV) provides rapid and complete protection from lethal challenge with the PRV variant emerging in Bartha-K61-vaccinated swine population in China. Vaccine 2014, 32, 3379–3385. [Google Scholar] [CrossRef]
- Dingwell, K.S.; Johnson, D.C. The herpes simplex virus gE-gI complex facilitates cell-to-cell spread and binds to components of cell junctions. J. Virol. 1998, 72, 8933–8942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skalsky, R.L.; Cullen, B.R. Viruses, microRNAs, and host interactions. Annu. Rev. Microbiol. 2010, 64, 123–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernier, A.; Sagan, S.M. The diverse roles of microRNAs at the host-virus interface. Viruses 2018, 10, 440. [Google Scholar] [CrossRef] [Green Version]
- Mahjoub, N.; Dhorne-Pollet, S.; Fuchs, W.; Endale Ahanda, M.L.; Lange, E.; Klupp, B.; Arya, A.; Loveland, J.E.; Lefevre, F.; Mettenleiter, T.C.; et al. A 2.5-kilobase deletion containing a cluster of nine microRNAs in the latency-associated-transcript locus of the pseudorabies virus affects the host response of porcine trigeminal ganglia during established latency. J. Virol. 2015, 89, 428–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contrant, M.; Fender, A.; Chane-Woon-Ming, B.; Randrianjafy, R.; Vivet-Boudou, V.; Richer, D.; Pfeffer, S. Importance of the RNA secondary structure for the relative accumulation of clustered viral microRNAs. Nucleic Acids Res. 2014, 42, 7981–7996. [Google Scholar] [CrossRef] [Green Version]
- Mettenleiter, T.C. Aujeszky’s disease (pseudorabies) virus: The virus and molecular pathogenesis—State of the art, June 1999. Vet. Res. 2000, 31, 99–115. [Google Scholar] [CrossRef] [Green Version]
- Valinezhad Orang, A.; Safaralizadeh, R.; Kazemzadeh-Bavili, M. Mechanisms of miRNA-mediated gene regulation from common downregulation to mRNA-specific upregulation. Int. J. Genom. 2014, 2014, 970607. [Google Scholar] [CrossRef] [Green Version]
- Brack, A.R.; Dijkstra, J.M.; Granzow, H.; Klupp, B.G.; Mettenleiter, T.C. Inhibition of virion maturation by simultaneous deletion of glycoproteins E, I, and M of pseudorabies virus. J. Virol. 1999, 73, 5364–5372. [Google Scholar] [CrossRef] [Green Version]
- Lamote, J.; Kestens, M.; Van Waesberghe, C.; Delva, J.; De Pelsmaeker, S.; Devriendt, B.; Favoreel, H.W. The pseudorabies virus glycoprotein gE/gI complex suppresses type I interferon production by plasmacytoid dendritic cells. J. Virol. 2017, 91, e02276-16. [Google Scholar] [CrossRef] [Green Version]


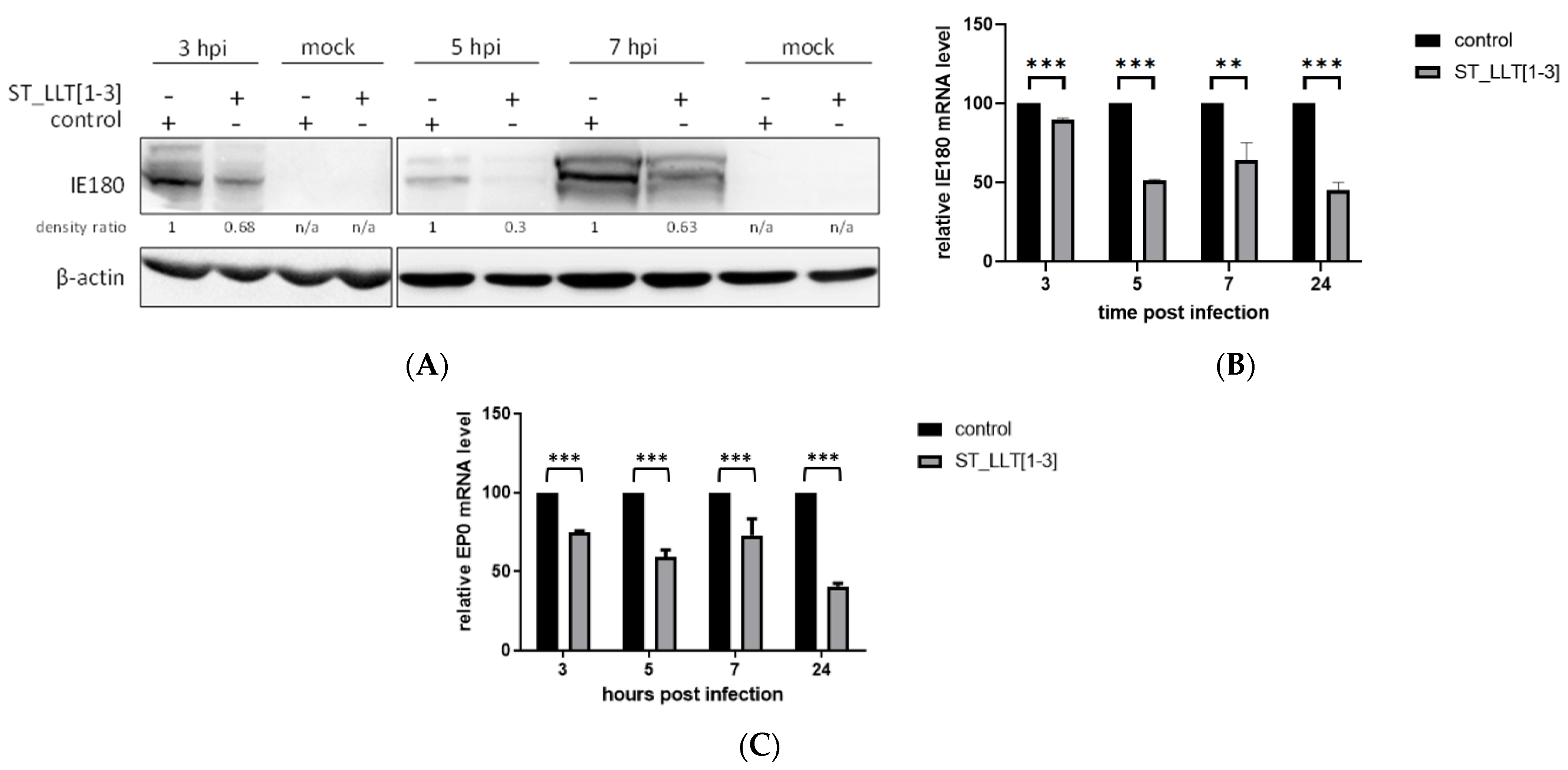



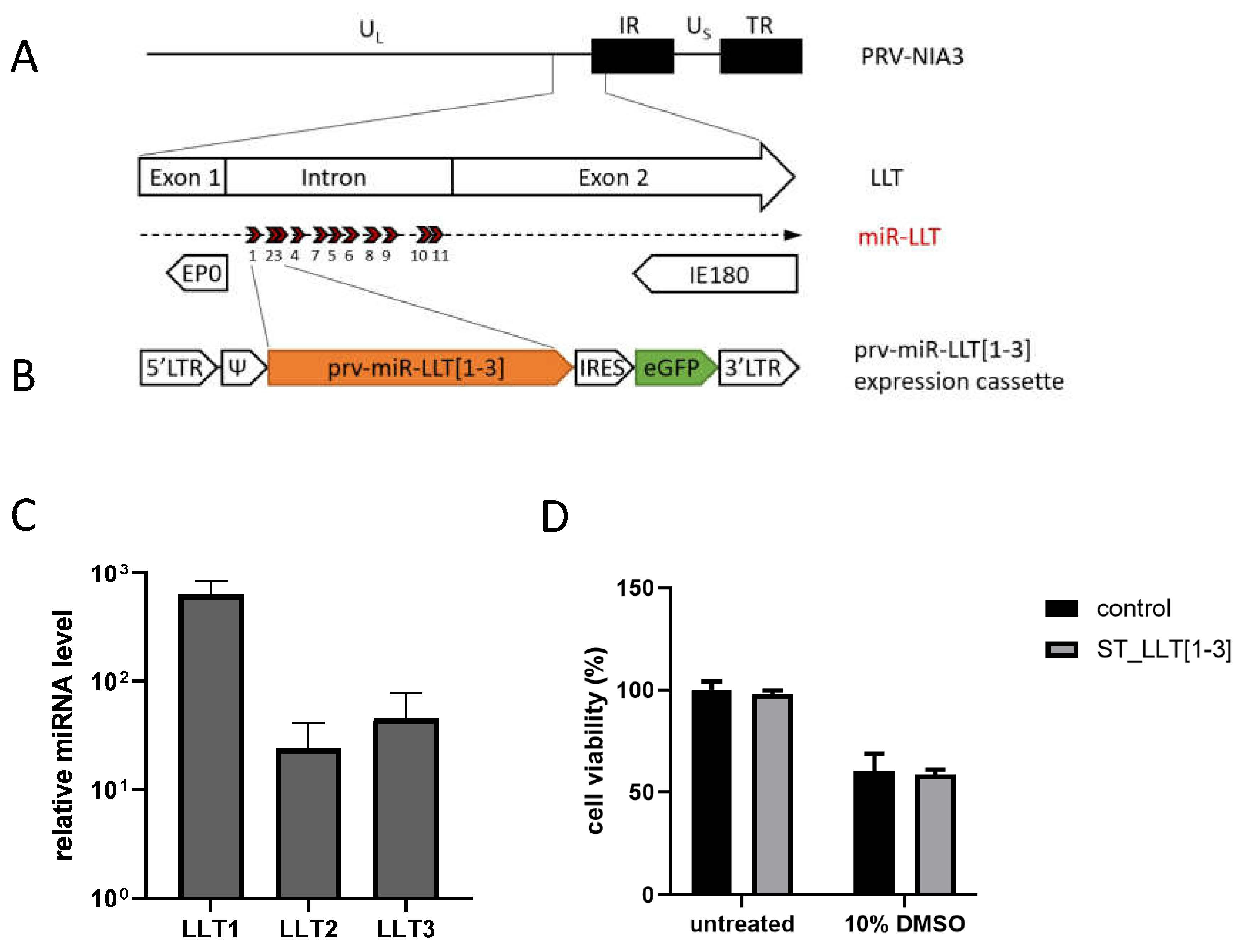{kind=link}
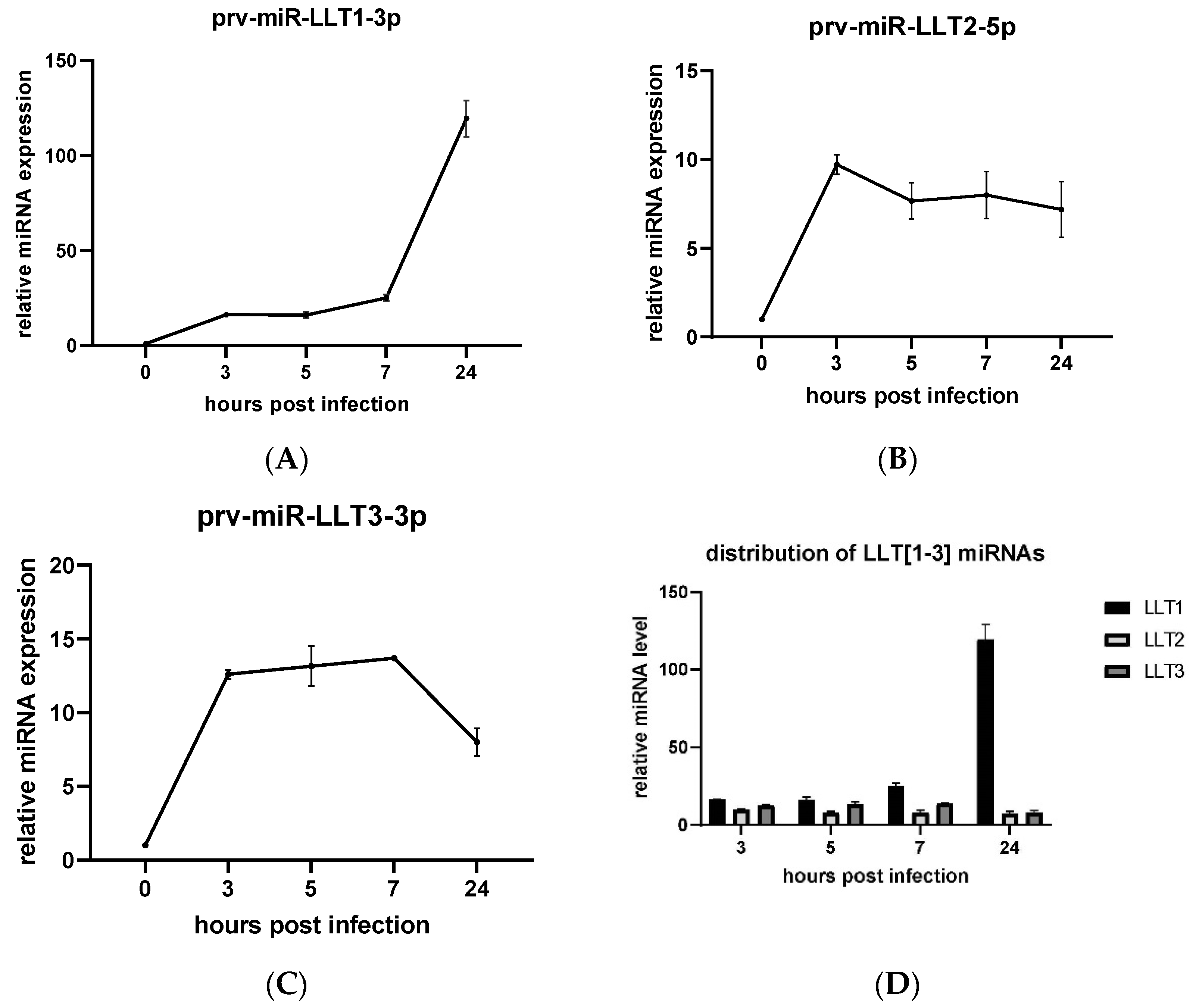{kind=link}
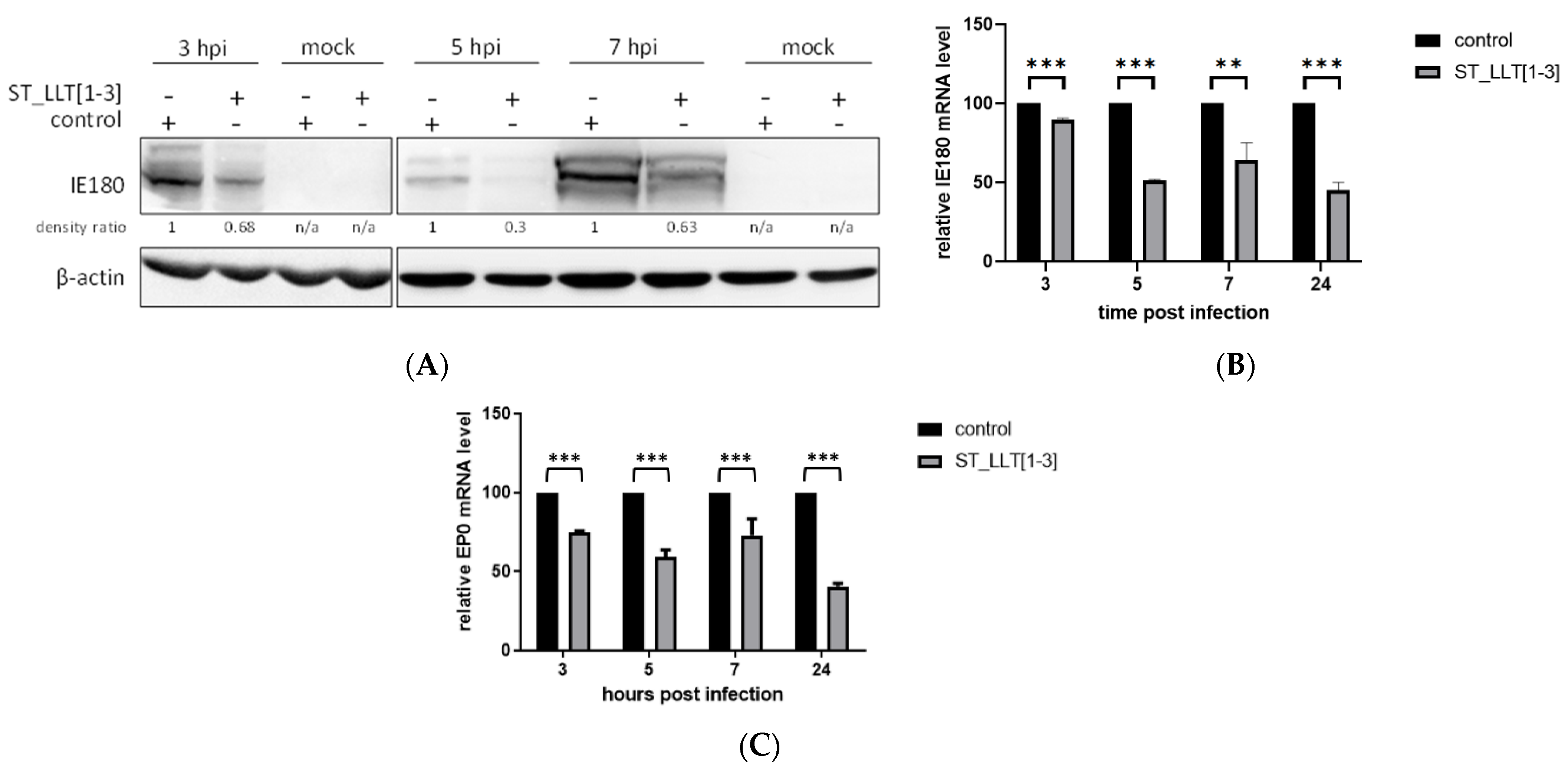{kind=link}
{kind=link}
{kind=link}
| Usage | Primer Name | Sequence (5′→3′); Restriction Enzymes Recognition Sites in Bold | Location (nt) in KU900059 |
|---|---|---|---|
| ST_LLT [1–3] construction | PRVmiR1-3BEF | AATGGATCCAGTCCATGACGGTGAGTG | 97,486–97,503 |
| PRVmiR1-3BER | GACGAATTCACGCTGATACTCTTGGCC | 98,670–98,687 | |
| miRNA qPCR | qPCR-F-miR-1 | TCTCACCCCTGGGTCCGTCGCAA | 97,837–97,857 |
| qPCR-F-miR-2 | CTCATCCCGTCAGACCTGCGAA | 98,298–98,317 | |
| qPCR-F-miR-3 | CGCACACGCCCCTCTCGCGCACAA | 98,398–98,419 | |
| mRNA qPCR(sequences from [32]) | RT2ie180F | CATCGTGCTGGACACCATCGAG | 103,891–103,912 |
| RT2ie180R | ACGTAGACGTGGTAGTCCCCCA | 103,844–103,865 | |
| RT2ep0F | GGGTGTGAACTATATCGACACGTC | 96,891–96,914 | |
| RT2ep0R | TCAGAGTCAGAGTGTGCCTCG | 96,864–96,884 | |
| RT2us8F | CTTCGACGTCTGGTTCCGC | 124,454–124,472 | |
| RT2us8R | GGTCACGCCATAGTTGGGC | 124,502–124,520 | |
| RT2s28F | GGGCCGAAACGATCTCAACC | n/a | |
| RT2s28R | GCCGGGCTTCTTACCCATT | n/a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffmann, W.; Lipińska, A.D.; Bieńkowska-Szewczyk, K. Functional Analysis of a Frontal miRNA Cluster Located in the Large Latency Transcript of Pseudorabies Virus. Viruses 2022, 14, 1147. https://doi.org/10.3390/v14061147
Hoffmann W, Lipińska AD, Bieńkowska-Szewczyk K. Functional Analysis of a Frontal miRNA Cluster Located in the Large Latency Transcript of Pseudorabies Virus. Viruses. 2022; 14(6):1147. https://doi.org/10.3390/v14061147
Chicago/Turabian StyleHoffmann, Weronika, Andrea D. Lipińska, and Krystyna Bieńkowska-Szewczyk. 2022. "Functional Analysis of a Frontal miRNA Cluster Located in the Large Latency Transcript of Pseudorabies Virus" Viruses 14, no. 6: 1147. https://doi.org/10.3390/v14061147