
Beyond Inhibition: A Novel Strategy of Targeting HIV-1 Protease to Eliminate Viral Reservoirs

{kind=link}
Abstract
:1. Introduction
2. HIV Protease Structure, Function, and Inhibitors
3. Host Cell Substrates of HIV-1 PR
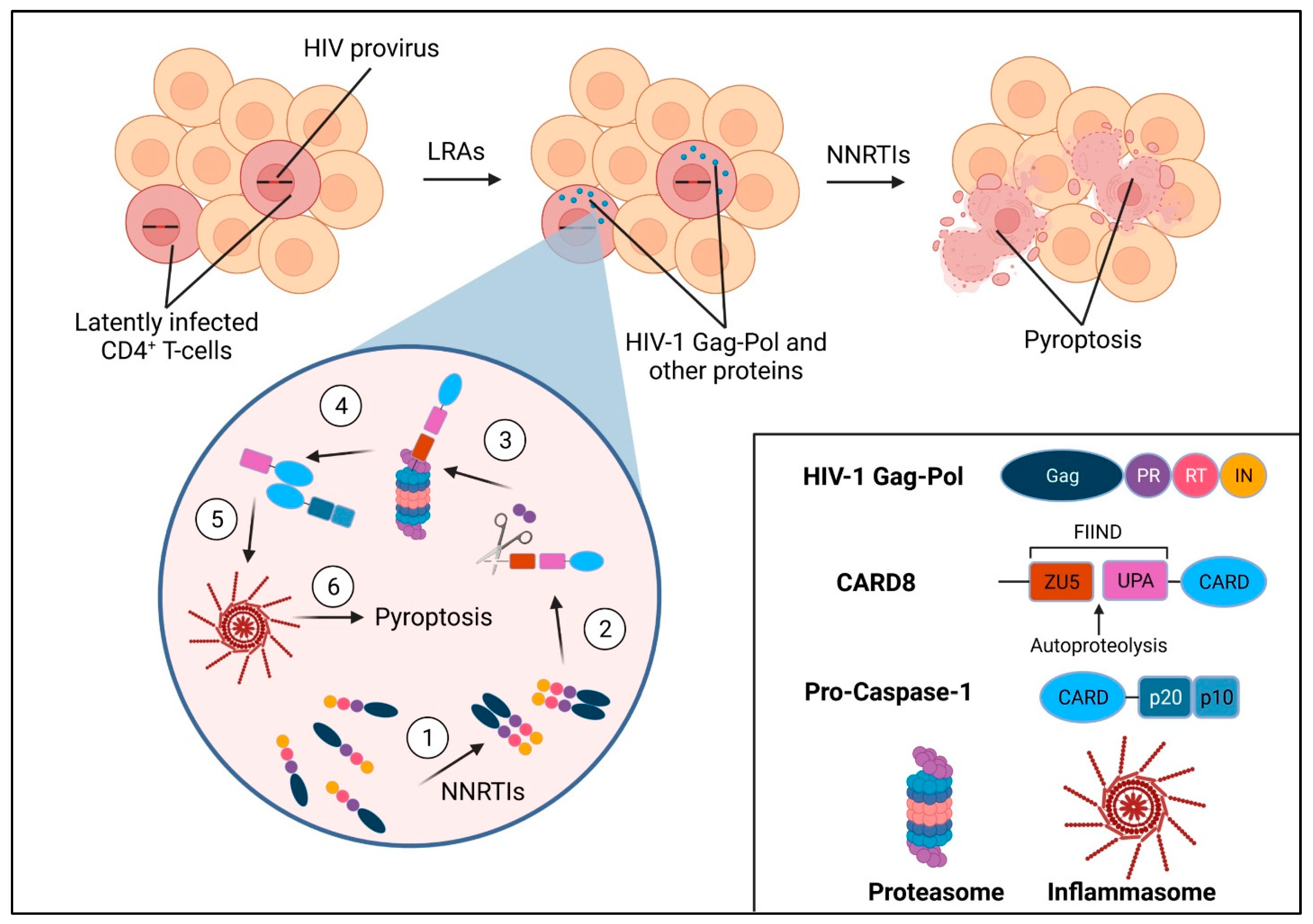
4. HIV-1 PR and the CARD8 Inflammasome
5. Conclusions and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Word Health Organization. HIV/AIDS. Available online: https://www.who.int/news-room/fact-sheets/detail/hiv-aids (accessed on 24 March 2022).
- Barre-Sinoussi, F.; Chermann, J.C.; Rey, F.; Nugeyre, M.T.; Chamaret, S.; Gruest, J.; Dauguet, C.; Axler-Blin, C.; Vezinet-Brun, F.; Rouzioux, C.; et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983, 220, 868–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallo, R.C.; Salahuddin, S.Z.; Popovic, M.; Shearer, G.M.; Kaplan, M.; Haynes, B.F.; Palker, T.J.; Redfield, R.; Oleske, J.; Safai, B.; et al. Frequent detection and isolation of cytopathic retroviruses (HTLV-III) from patients with AIDS and at risk for AIDS. Science 1984, 224, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.A.; Hoffman, A.D.; Kramer, S.M.; Landis, J.A.; Shimabukuro, J.M.; Oshiro, L.S. Isolation of lymphocytopathic retroviruses from San Francisco patients with AIDS. Science 1984, 225, 840–842. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.; Cherepanov, P. The structural biology of HIV-1: Mechanistic and therapeutic insights. Nat. Rev. Microbiol. 2012, 10, 279–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, D.D.; Neumann, A.U.; Perelson, A.S.; Chen, W.; Leonard, J.M.; Markowitz, M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 1995, 373, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Palella, F.J., Jr.; Delaney, K.M.; Moorman, A.C.; Loveless, M.O.; Fuhrer, J.; Satten, G.A.; Aschman, D.J.; Holmberg, S.D. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N. Engl. J. Med. 1998, 338, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Fauci, A.S. Latent reservoirs of HIV: Obstacles to the eradication of virus. Proc. Natl. Acad. Sci. USA 1999, 96, 10958–10961. [Google Scholar] [CrossRef] [Green Version]
- Chun, T.W.; Stuyver, L.; Mizell, S.B.; Ehler, L.A.; Mican, J.A.; Baseler, M.; Lloyd, A.L.; Nowak, M.A.; Fauci, A.S. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 1997, 94, 13193–13197. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.K.; Hezareh, M.; Gunthard, H.F.; Havlir, D.V.; Ignacio, C.C.; Spina, C.A.; Richman, D.D. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 1997, 278, 1291–1295. [Google Scholar] [CrossRef]
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef]
- Konvalinka, J.; Krausslich, H.G.; Muller, B. Retroviral proteases and their roles in virion maturation. Virology 2015, 479–480, 403–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohl, N.E.; Emini, E.A.; Schleif, W.A.; Davis, L.J.; Heimbach, J.C.; Dixon, R.A.; Scolnick, E.M.; Sigal, I.S. Active human immunodeficiency virus protease is required for viral infectivity. Proc. Natl. Acad. Sci. USA 1988, 85, 4686–4690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Word Health Organization. FDA-Approved HIV Medicines. Available online: https://hivinfo.nih.gov/understanding-hiv/fact-sheets/fda-approved-hiv-medicines (accessed on 26 March 2022).
- Impens, F.; Timmerman, E.; Staes, A.; Moens, K.; Arien, K.K.; Verhasselt, B.; Vandekerckhove, J.; Gevaert, K. A catalogue of putative HIV-1 protease host cell substrates. Biol. Chem. 2012, 393, 915–931. [Google Scholar] [CrossRef] [PubMed]
- Ventoso, I.; Blanco, R.; Perales, C.; Carrasco, L. HIV-1 protease cleaves eukaryotic initiation factor 4G and inhibits cap-dependent translation. Proc. Natl. Acad. Sci. USA 2001, 98, 12966–12971. [Google Scholar] [CrossRef] [Green Version]
- Strack, P.R.; Frey, M.W.; Rizzo, C.J.; Cordova, B.; George, H.J.; Meade, R.; Ho, S.P.; Corman, J.; Tritch, R.; Korant, B.D. Apoptosis mediated by HIV protease is preceded by cleavage of Bcl-2. Proc. Natl. Acad. Sci. USA 1996, 93, 9571–9576. [Google Scholar] [CrossRef] [Green Version]
- Nie, Z.; Phenix, B.N.; Lum, J.J.; Alam, A.; Lynch, D.H.; Beckett, B.; Krammer, P.H.; Sekaly, R.P.; Badley, A.D. HIV-1 protease processes procaspase 8 to cause mitochondrial release of cytochrome c, caspase cleavage and nuclear fragmentation. Cell Death Differ. 2002, 9, 1172–1184. [Google Scholar] [CrossRef]
- Wang, Q.; Gao, H.; Clark, K.M.; Mugisha, C.S.; Davis, K.; Tang, J.P.; Harlan, G.H.; DeSelm, C.J.; Presti, R.M.; Kutluay, S.B.; et al. CARD8 is an inflammasome sensor for HIV-1 protease activity. Science 2021, 371, eabe1707. [Google Scholar] [CrossRef]
- Lv, Z.; Chu, Y.; Wang, Y. HIV protease inhibitors: A review of molecular selectivity and toxicity. HIV AIDS 2015, 7, 95–104. [Google Scholar] [CrossRef] [Green Version]
- Pearl, L.H.; Taylor, W.R. A structural model for the retroviral proteases. Nature 1987, 329, 351–354. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Osswald, H.L.; Prato, G. Recent Progress in the Development of HIV-1 Protease Inhibitors for the Treatment of HIV/AIDS. J. Med. Chem. 2016, 59, 5172–5208. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, L.K.; Yamazaki, T.; Torchia, D.A.; Grzesiek, S.; Bax, A.; Stahl, S.J.; Kaufman, J.D.; Wingfield, P.T.; Lam, P.Y.; Jadhav, P.K.; et al. Flexibility and function in HIV-1 protease. Nat. Struct. Biol. 1995, 2, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Prabu-Jeyabalan, M.; Nalivaika, E.; Schiffer, C.A. Substrate shape determines specificity of recognition for HIV-1 protease: Analysis of crystal structures of six substrate complexes. Structure 2002, 10, 369–381. [Google Scholar] [CrossRef]
- Wensing, A.M.; van Maarseveen, N.M.; Nijhuis, M. Fifteen years of HIV Protease Inhibitors: Raising the barrier to resistance. Antiviral Res. 2010, 85, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Wiegers, K.; Rutter, G.; Kottler, H.; Tessmer, U.; Hohenberg, H.; Krausslich, H.G. Sequential steps in human immunodeficiency virus particle maturation revealed by alterations of individual Gag polyprotein cleavage sites. J. Virol. 1998, 72, 2846–2854. [Google Scholar] [CrossRef] [Green Version]
- Krausslich, H.G. Human immunodeficiency virus proteinase dimer as component of the viral polyprotein prevents particle assembly and viral infectivity. Proc. Natl. Acad. Sci. USA 1991, 88, 3213–3217. [Google Scholar] [CrossRef] [Green Version]
- Wyma, D.J.; Jiang, J.; Shi, J.; Zhou, J.; Lineberger, J.E.; Miller, M.D.; Aiken, C. Coupling of human immunodeficiency virus type 1 fusion to virion maturation: A novel role of the gp41 cytoplasmic tail. J. Virol. 2004, 78, 3429–3435. [Google Scholar] [CrossRef] [Green Version]
- Murakami, T.; Ablan, S.; Freed, E.O.; Tanaka, Y. Regulation of human immunodeficiency virus type 1 Env-mediated membrane fusion by viral protease activity. J. Virol. 2004, 78, 1026–1031. [Google Scholar] [CrossRef] [Green Version]
- Louis, J.M.; Clore, G.M.; Gronenborn, A.M. Autoprocessing of HIV-1 protease is tightly coupled to protein folding. Nat. Struct. Biol. 1999, 6, 868–875. [Google Scholar] [CrossRef]
- Tabler, C.O.; Wegman, S.J.; Chen, J.; Shroff, H.; Alhusaini, N.; Tilton, J.C. The HIV-1 Viral Protease Is Activated during Assembly and Budding Prior to Particle Release. J. Virol. 2022, 96, e0219821. [Google Scholar] [CrossRef]
- Wlodawer, A.; Erickson, J.W. Structure-based inhibitors of HIV-1 protease. Annu. Rev. Biochem. 1993, 62, 543–585. [Google Scholar] [CrossRef]
- Hammer, S.M.; Squires, K.E.; Hughes, M.D.; Grimes, J.M.; Demeter, L.M.; Currier, J.S.; Eron, J.J., Jr.; Feinberg, J.E.; Balfour, H.H., Jr.; Deyton, L.R.; et al. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. AIDS Clinical Trials Group 320 Study Team. N. Engl. J. Med. 1997, 337, 725–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulick, R.M.; Mellors, J.W.; Havlir, D.; Eron, J.J.; Gonzalez, C.; McMahon, D.; Richman, D.D.; Valentine, F.T.; Jonas, L.; Meibohm, A.; et al. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N. Engl. J. Med. 1997, 337, 734–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, I.T.; Wang, Y.F.; Harrison, R.W. HIV Protease: Historical Perspective and Current Research. Viruses 2021, 13, 839. [Google Scholar] [CrossRef] [PubMed]
- Kempf, D.J.; Marsh, K.C.; Kumar, G.; Rodrigues, A.D.; Denissen, J.F.; McDonald, E.; Kukulka, M.J.; Hsu, A.; Granneman, G.R.; Baroldi, P.A.; et al. Pharmacokinetic enhancement of inhibitors of the human immunodeficiency virus protease by coadministration with ritonavir. Antimicrob. Agents Chemother. 1997, 41, 654–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoeman, R.L.; Huttermann, C.; Hartig, R.; Traub, P. Amino-terminal polypeptides of vimentin are responsible for the changes in nuclear architecture associated with human immunodeficiency virus type 1 protease activity in tissue culture cells. Mol. Biol. Cell 2001, 12, 143–154. [Google Scholar] [CrossRef] [Green Version]
- Shoeman, R.L.; Honer, B.; Stoller, T.J.; Kesselmeier, C.; Miedel, M.C.; Traub, P.; Graves, M.C. Human immunodeficiency virus type 1 protease cleaves the intermediate filament proteins vimentin, desmin, and glial fibrillary acidic protein. Proc. Natl. Acad. Sci. USA 1990, 87, 6336–6340. [Google Scholar] [CrossRef] [Green Version]
- Jager, S.; Cimermancic, P.; Gulbahce, N.; Johnson, J.R.; McGovern, K.E.; Clarke, S.C.; Shales, M.; Mercenne, G.; Pache, L.; Li, K.; et al. Global landscape of HIV-human protein complexes. Nature 2011, 481, 365–370. [Google Scholar] [CrossRef]
- Gomes-Duarte, A.; Lacerda, R.; Menezes, J.; Romao, L. eIF3: A factor for human health and disease. RNA Biol. 2018, 15, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Vaux, D.L.; Cory, S.; Adams, J.M. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 1988, 335, 440–442. [Google Scholar] [CrossRef]
- Li, H.; Zhu, H.; Xu, C.J.; Yuan, J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Wei, M.C.; Lindsten, T.; Mootha, V.K.; Weiler, S.; Gross, A.; Ashiya, M.; Thompson, C.B.; Korsmeyer, S.J. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000, 14, 2060–2071. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Bren, G.D.; Vlahakis, S.R.; Schimnich, A.A.; Brenchley, J.M.; Trushin, S.A.; Warren, S.; Schnepple, D.J.; Kovacs, C.M.; Loutfy, M.R.; et al. Human immunodeficiency virus type 1 protease cleaves procaspase 8 in vivo. J. Virol. 2007, 81, 6947–6956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurczyszak, D.; Zhang, W.; Terry, S.N.; Kehrer, T.; Bermudez Gonzalez, M.C.; McGregor, E.; Mulder, L.C.F.; Eckwahl, M.J.; Pan, T.; Simon, V. HIV protease cleaves the antiviral m6A reader protein YTHDF3 in the viral particle. PLoS Pathog. 2020, 16, e1008305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastan, J.P.; Tremblay, M.W.; Brown, M.C.; Trimarco, J.D.; Dobrikova, E.Y.; Dobrikov, M.I.; Gromeier, M. Enterovirus 2A(pro) Cleavage of the YTHDF m(6)A Readers Implicates YTHDF3 as a Mediator of Type I Interferon-Driven JAK/STAT Signaling. mBio 2021, 12, e00116-21. [Google Scholar] [CrossRef] [PubMed]
- Eng, V.V.; Wemyss, M.A.; Pearson, J.S. The diverse roles of RIP kinases in host-pathogen interactions. Semin. Cell Dev. Biol. 2021, 109, 125–143. [Google Scholar] [CrossRef]
- Wagner, R.N.; Reed, J.C.; Chanda, S.K. HIV-1 protease cleaves the serine-threonine kinases RIPK1 and RIPK2. Retrovirology 2015, 12, 74. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, W.J.; Upton, J.W.; Mocarski, E.S. Viral modulation of programmed necrosis. Curr. Opin. Virol. 2013, 3, 296–306. [Google Scholar] [CrossRef] [Green Version]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 2012, 11, 290–297. [Google Scholar] [CrossRef] [Green Version]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Taabazuing, C.Y.; Griswold, A.R.; Bachovchin, D.A. The NLRP1 and CARD8 inflammasomes. Immunol. Rev. 2020, 297, 13–25. [Google Scholar] [CrossRef]
- Broz, P.; von Moltke, J.; Jones, J.W.; Vance, R.E.; Monack, D.M. Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe 2010, 8, 471–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poyet, J.L.; Srinivasula, S.M.; Tnani, M.; Razmara, M.; Fernandes-Alnemri, T.; Alnemri, E.S. Identification of Ipaf, a human caspase-1-activating protein related to Apaf-1. J. Biol. Chem. 2001, 276, 28309–28313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broz, P.; Pelegrin, P.; Shao, F. The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 2020, 20, 143–157. [Google Scholar] [CrossRef]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef]
- Fink, S.L.; Cookson, B.T. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 2006, 8, 1812–1825. [Google Scholar] [CrossRef]
- Jorgensen, I.; Miao, E.A. Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev. 2015, 265, 130–142. [Google Scholar] [CrossRef]
- Razmara, M.; Srinivasula, S.M.; Wang, L.; Poyet, J.L.; Geddes, B.J.; DiStefano, P.S.; Bertin, J.; Alnemri, E.S. CARD-8 protein, a new CARD family member that regulates caspase-1 activation and apoptosis. J. Biol. Chem. 2002, 277, 13952–13958. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.C.; Taabazuing, C.Y.; Okondo, M.C.; Chui, A.J.; Rao, S.D.; Brown, F.C.; Reed, C.; Peguero, E.; de Stanchina, E.; Kentsis, A.; et al. DPP8/DPP9 inhibitor-induced pyroptosis for treatment of acute myeloid leukemia. Nat. Med. 2018, 24, 1151–1156. [Google Scholar] [CrossRef]
- D’Osualdo, A.; Weichenberger, C.X.; Wagner, R.N.; Godzik, A.; Wooley, J.; Reed, J.C. CARD8 and NLRP1 undergo autoproteolytic processing through a ZU5-like domain. PLoS ONE 2011, 6, e27396. [Google Scholar] [CrossRef] [Green Version]
- Finger, J.N.; Lich, J.D.; Dare, L.C.; Cook, M.N.; Brown, K.K.; Duraiswami, C.; Bertin, J.; Gough, P.J. Autolytic proteolysis within the function to find domain (FIIND) is required for NLRP1 inflammasome activity. J. Biol. Chem. 2012, 287, 25030–25037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, K.S.; Teo, D.E.T.; Tan, K.S.; Toh, G.A.; Ong, H.H.; Lim, C.K.; Lay, K.; Au, B.V.; Lew, T.S.; Chu, J.J.H.; et al. Enteroviral 3C protease activates the human NLRP1 inflammasome in airway epithelia. Science 2020, 370, eaay2002. [Google Scholar] [CrossRef] [PubMed]
- Bauernfried, S.; Scherr, M.J.; Pichlmair, A.; Duderstadt, K.E.; Hornung, V. Human NLRP1 is a sensor for double-stranded RNA. Science 2021, 371, eabd0811. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, A.; Moore, K.L.; Mak, J.; Sluis-Cremer, N.; de Bethune, M.P.; Tachedjian, G. Potent nonnucleoside reverse transcriptase inhibitors target HIV-1 Gag-Pol. PLoS Pathog. 2006, 2, e119. [Google Scholar] [CrossRef] [Green Version]
- Phillips, R.E.; Rowland-Jones, S.; Nixon, D.F.; Gotch, F.M.; Edwards, J.P.; Ogunlesi, A.O.; Elvin, J.G.; Rothbard, J.A.; Bangham, C.R.; Rizza, C.R.; et al. Human immunodeficiency virus genetic variation that can escape cytotoxic T cell recognition. Nature 1991, 354, 453–459. [Google Scholar] [CrossRef]
- Caskey, M.; Klein, F.; Lorenzi, J.C.; Seaman, M.S.; West, A.P., Jr.; Buckley, N.; Kremer, G.; Nogueira, L.; Braunschweig, M.; Scheid, J.F.; et al. Viraemia suppressed in HIV-1-infected humans by broadly neutralizing antibody 3BNC117. Nature 2015, 522, 487–491. [Google Scholar] [CrossRef] [Green Version]
- Kwong, P.D.; Doyle, M.L.; Casper, D.J.; Cicala, C.; Leavitt, S.A.; Majeed, S.; Steenbeke, T.D.; Venturi, M.; Chaiken, I.; Fung, M.; et al. HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature 2002, 420, 678–682. [Google Scholar] [CrossRef]
- Dinarello, C.A. A clinical perspective of IL-1beta as the gatekeeper of inflammation. Eur. J. Immunol. 2011, 41, 1203–1217. [Google Scholar] [CrossRef]
- Johnson, D.C.; Okondo, M.C.; Orth, E.L.; Rao, S.D.; Huang, H.C.; Ball, D.P.; Bachovchin, D.A. DPP8/9 inhibitors activate the CARD8 inflammasome in resting lymphocytes. Cell Death Dis. 2020, 11, 628. [Google Scholar] [CrossRef]
- Massanella, M.; Richman, D.D. Measuring the latent reservoir in vivo. J. Clin. Investig. 2016, 126, 464–472. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.E.; Jaworowski, A.; Hearps, A.C. The HIV Reservoir in Monocytes and Macrophages. Front. Immunol. 2019, 10, 1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boffito, M.; Back, D.J.; Blaschke, T.F.; Rowland, M.; Bertz, R.J.; Gerber, J.G.; Miller, V. Protein binding in antiretroviral therapies. AIDS Res. Hum. Retrovir. 2003, 19, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.; Wang, Q.; Shan, L. CARD8 inflammasome sensitization through DPP9 inhibition enhances NNRTI-triggered killing of HIV-1-infected cells. bioRxiv, 2021; Preprint. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.G.; Shan, L. Beyond Inhibition: A Novel Strategy of Targeting HIV-1 Protease to Eliminate Viral Reservoirs. Viruses 2022, 14, 1179. https://doi.org/10.3390/v14061179
Kim JG, Shan L. Beyond Inhibition: A Novel Strategy of Targeting HIV-1 Protease to Eliminate Viral Reservoirs. Viruses. 2022; 14(6):1179. https://doi.org/10.3390/v14061179
Chicago/Turabian StyleKim, Josh G., and Liang Shan. 2022. "Beyond Inhibition: A Novel Strategy of Targeting HIV-1 Protease to Eliminate Viral Reservoirs" Viruses 14, no. 6: 1179. https://doi.org/10.3390/v14061179
APA StyleKim, J. G., & Shan, L. (2022). Beyond Inhibition: A Novel Strategy of Targeting HIV-1 Protease to Eliminate Viral Reservoirs. Viruses, 14(6), 1179. https://doi.org/10.3390/v14061179