
Repurposing an In Vitro Measles Virus Dissemination Assay for Screening of Antiviral Compounds

 , ,
, ,  and
and
Abstract
:1. Introduction
1.1. Measles Virus
1.2. Pathogenesis
1.3. Treatment Challenges
1.4. In Vitro Testing
2. Materials and Methods
2.1. Cells and Viruses
2.2. Antiviral Compounds
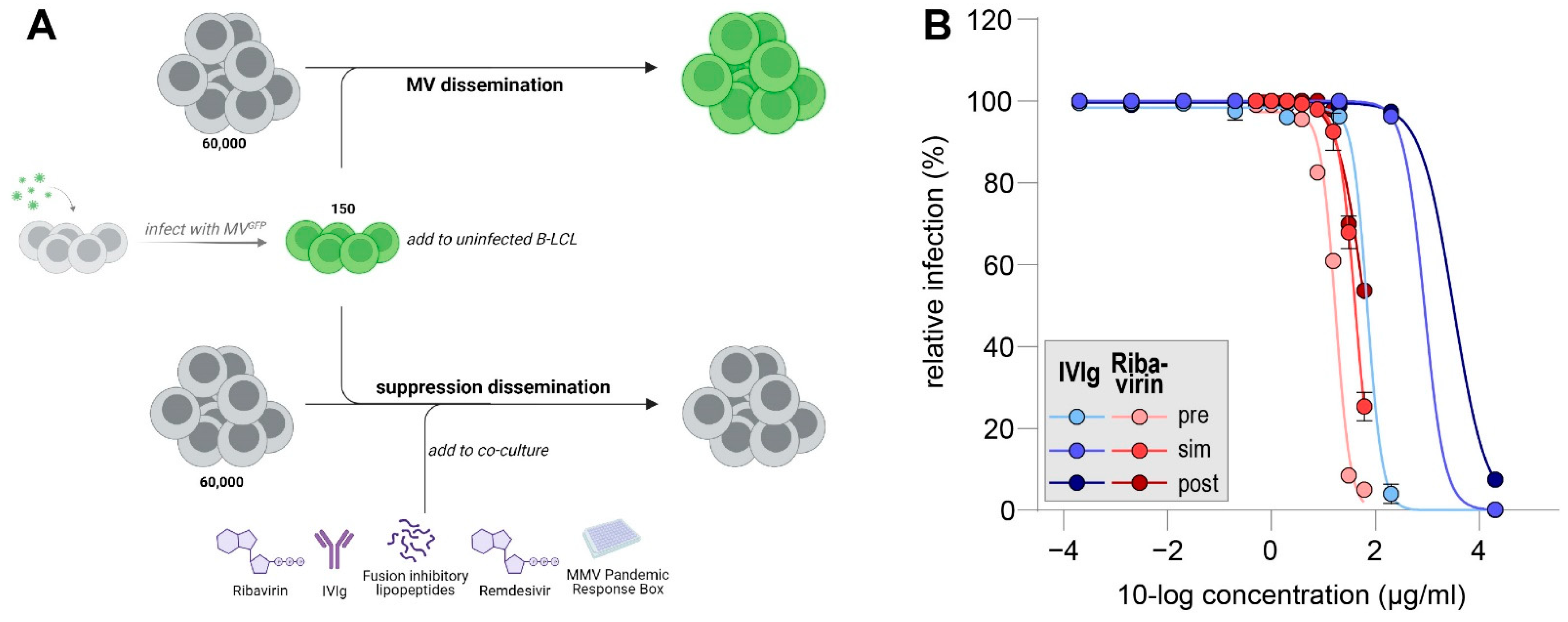
2.3. In Vitro MV Dissemination Assay
2.4. Data Analysis
3. Results
3.1. IVIg Inhibitis MV-Dissemination at Physiological Concentrations
3.2. Remdesivir Efficiently Inhibitis MV-Dissemination at Sub-Micromolar Concentrations
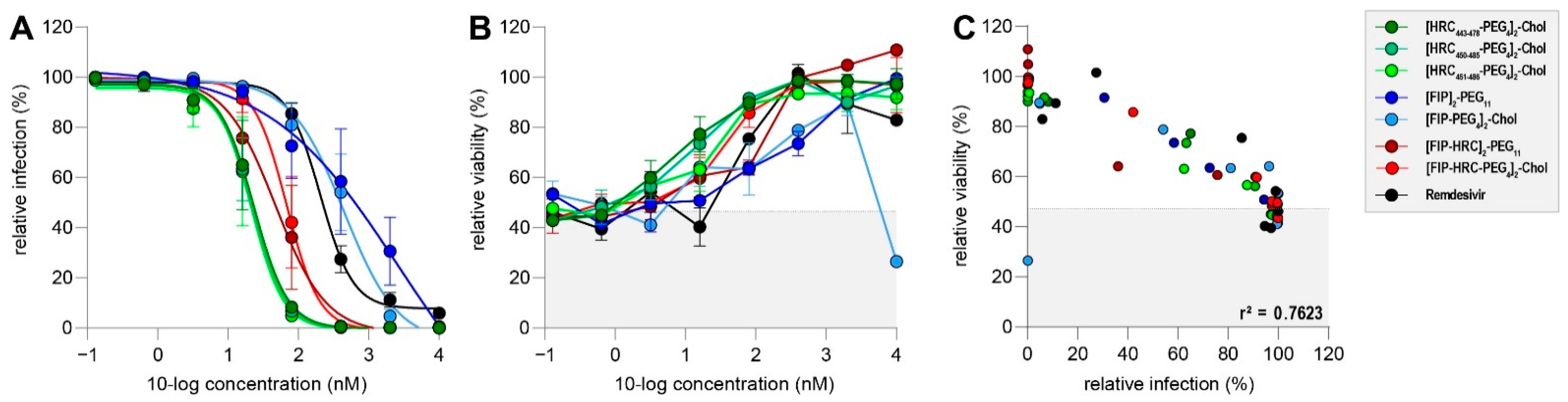
3.3. Fusion Inhibitory Lipopeptides Prevent MV Dissemination at Nanomolar Concentrations
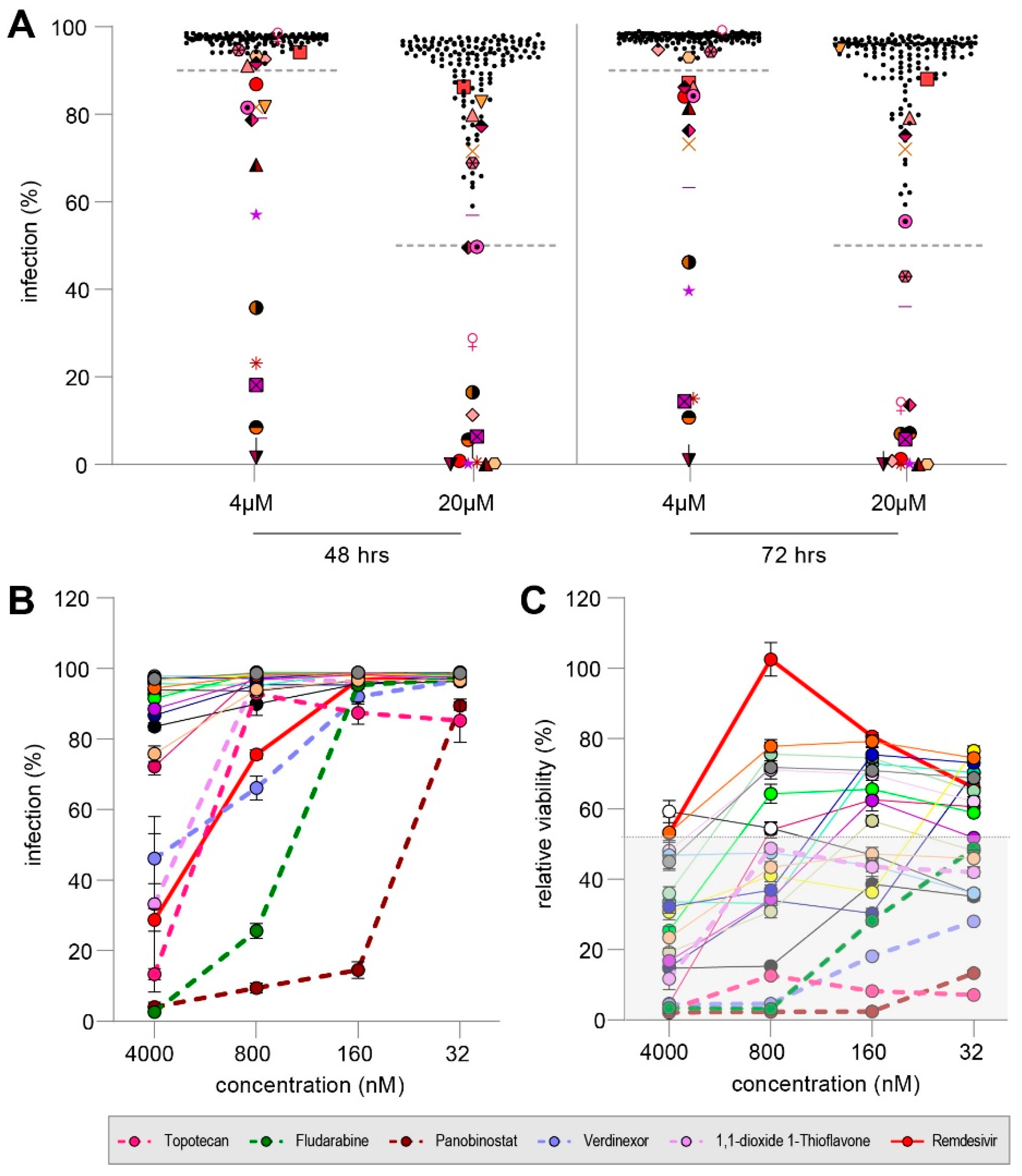
3.4. MMV Pandemic Response Box Screening
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Measles & Rubella Initiative. Measles & Rubella Strategic Framework 2021—2030. Available online: https://measlesrubellainitiative.org/measles-rubella-strategic-framework-2021-2030/ (accessed on 20 February 2022).
- WHO. Measles Fact Sheet. Available online: https://www.who.int/news-room/fact-sheets/detail/measles (accessed on 20 February 2022).
- WHO. Worldwide Measles Deaths Climb 50% from 2016 to 2019 Claiming over 207,500 Lives in 2019. Available online: https://www.who.int/news/item/12-11-2020-worldwide-measles-deaths-climb-50-from-2016-to-2019-claiming-over-207-500-lives-in-2019 (accessed on 20 February 2022).
- Dixon, M.G.; Ferrari, M.; Antoni, S.; Li, X.; Portnoy, A.; Lambert, B.; Hauryski, S.; Hatcher, C.; Nedelec, Y.; Patel, M.; et al. Progress toward Regional Measles Elimination—Worldwide, 2000–2020. Morb. Mortal Wkly. Rep. 2021, 70, 1563–1569. [Google Scholar] [CrossRef] [PubMed]
- Torjesen, I. Measles outbreaks likely as covid pandemic leaves millions of world’s children unvaccinated, WHO warns. BMJ 2021, 375, n2755. [Google Scholar] [CrossRef] [PubMed]
- Rima, B.; Balkema-Buschmann, A.; Dundon, W.G.; Duprex, P.; Easton, A.; Fouchier, R.; Kurath, G.; Lamb, R.; Lee, B.; Rota, P.; et al. ICTV Virus Taxonomy Profile: Paramyxoviridae. J. Gen. Virol. 2019, 100, 1593–1594. [Google Scholar] [CrossRef] [PubMed]
- WHO. Genetic diversity of wild-type measles viruses and the global measles nucleotide surveillance database (MeaNS). Wkly. Epidemiol. Rec. 2015, 90, 373–380. [Google Scholar]
- Rota, P.A.; Brown, K.; Mankertz, A.; Santibanez, S.; Shulga, S.; Muller, C.P.; Hübschen, J.M.; Siqueira, M.; Beirnes, J.; Ahmed, H.; et al. Global Distribution of Measles Genotypes and Measles Molecular Epidemiology. J. Infect. Dis. 2011, 204, S514–S523. [Google Scholar] [CrossRef]
- Dowling, P.C.; Blumberg, B.M.; Menonna, J.; Adamus, J.E.; Cook, P.; Crowley, J.C.; Kolakofsky, D.; Cook, S.D. Transcriptional map of the measles virus genome. J. Gen. Virol. 1986, 67 Pt 9, 1987–1992. [Google Scholar] [CrossRef]
- Bankamp, B.; Horikami, S.M.; Thompson, P.D.; Huber, M.; Billeter, M.; Moyer, S.A. Domains of the Measles Virus N Protein Required for Binding to P Protein and Self-Assembly. Virology 1996, 216, 272–277. [Google Scholar] [CrossRef] [Green Version]
- Plattet, P.; Alves, L.; Herren, M.; Aguilar, H.C. Measles Virus Fusion Protein: Structure, Function and Inhibition. Viruses 2016, 8, 112. [Google Scholar] [CrossRef] [Green Version]
- Tatsuo, H.; Ono, N.; Yanagi, Y. Morbilliviruses Use Signaling Lymphocyte Activation Molecules (CD150) as Cellular Receptors. J. Virol. 2001, 75, 5842–5850. [Google Scholar] [CrossRef] [Green Version]
- Mühlebach, M.D.; Mateo, M.; Sinn, P.L.; Prüfer, S.; Uhlig, K.M.; Leonard, V.H.; Navaratnarajah, C.K.; Frenzke, M.; Wong, X.X.; Sawatsky, B.; et al. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature 2011, 480, 530–533. [Google Scholar] [CrossRef] [Green Version]
- Noyce, R.S.; Bondre, D.G.; Ha, M.N.; Lin, L.T.; Sisson, G.; Tsao, M.S.; Richardson, C.D. Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog. 2011, 7, e1002240. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, M.; Takeda, M.; Shirogane, Y.; Nakatsu, Y.; Nakamura, T.; Yanagi, Y. The Matrix Protein of Measles Virus Regulates Viral RNA Synthesis and Assembly by Interacting with the Nucleocapsid Protein. J. Virol. 2009, 83, 10374–10383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, R.M.; Plemper, R.K. Structure and organization of paramyxovirus particles. Curr. Opin. Virol. 2017, 24, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Liston, P.; DiFlumeri, C.; Briedis, D.J. Protein interactions entered into by the measles virus P, V, and C proteins. Virus Res. 1995, 38, 241–259. [Google Scholar] [CrossRef]
- Guerra, F.M.; Bolotin, S.; Lim, G.; Heffernan, J.; Deeks, S.L.; Li, Y.; Crowcroft, N.S. The basic reproduction number (R0) of measles: A systematic review. Lancet Infect. Dis. 2017, 17, e420–e428. [Google Scholar] [CrossRef]
- Lemon, K.; de Vries, R.D.; Mesman, A.W.; McQuaid, S.; van Amerongen, G.; Yüksel, S.; Ludlow, M.; Rennick, L.J.; Kuiken, T.; Rima, B.K.; et al. Early target cells of measles virus after aerosol infection of non-human primates. PLoS Pathog. 2011, 7, e1001263. [Google Scholar] [CrossRef]
- De Swart, R.L.; Ludlow, M.; de Witte, L.; Yanagi, Y.; van Amerongen, G.; McQuaid, S.; Yüksel, S.; Geijtenbeek, T.B.; Duprex, W.P.; Osterhaus, A.D. Predominant infection of CD150+ lymphocytes and dendritic cells during measles virus infection of macaques. PLoS Pathog. 2007, 3, e178. [Google Scholar] [CrossRef]
- De Vries, R.D.; Lemon, K.; Ludlow, M.; McQuaid, S.; Yüksel, S.; van Amerongen, G.; Rennick, L.J.; Rima, B.K.; Osterhaus, A.D.; de Swart, R.L. In vivo tropism of attenuated and pathogenic measles virus expressing green fluorescent protein in macaques. J. Virol. 2010, 84, 4714–4724. [Google Scholar] [CrossRef] [Green Version]
- Ludlow, M.; Rennick, L.J.; Sarlang, S.; Skibinski, G.; McQuaid, S.; Moore, T.; de Swart, R.L.; Duprex, W.P. Wild-type measles virus infection of primary epithelial cells occurs via the basolateral surface without syncytium formation or release of infectious virus. J. Gen. Virol. 2010, 91, 971–979. [Google Scholar] [CrossRef]
- Racaniello, V. An Exit Strategy for Measles Virus. Science 2011, 334, 1650–1651. [Google Scholar] [CrossRef]
- De Vries, R.D.; McQuaid, S.; van Amerongen, G.; Yüksel, S.; Verburgh, R.J.; Osterhaus, A.D.; Duprex, W.P.; de Swart, R.L. Measles immune suppression: Lessons from the macaque model. PLoS Pathog. 2012, 8, e1002885. [Google Scholar] [CrossRef] [PubMed]
- Mina, M.J.; Metcalf, C.J.E.; de Swart, R.L.; Osterhaus, A.D.M.E.; Grenfell, B.T. Long-term measles-induced immunomodulation increases overall childhood infectious disease mortality. Science 2015, 348, 694–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, S.E.; Yates, A.J.; de Swart, R.L.; de Vries, R.D.; Mina, M.J.; Nelson, A.N.; Lin, W.-H.W.; Kouyos, R.D.; Griffin, D.E.; Grenfell, B.T. Modeling the measles paradox reveals the importance of cellular immunity in regulating viral clearance. PLoS Pathog. 2018, 14, e1007493. [Google Scholar] [CrossRef] [PubMed]
- Laksono, B.M.; Fortugno, P.; Nijmeijer, B.M.; de Vries, R.D.; Cordisco, S.; Kuiken, T.; Geijtenbeek, T.B.H.; Duprex, W.P.; Brancati, F.; de Swart, R.L. Measles skin rash: Infection of lymphoid and myeloid cells in the dermis precedes viral dissemination to the epidermis. PLoS Pathog. 2020, 16, e1008253. [Google Scholar] [CrossRef]
- Ferren, M.; Horvat, B.; Mathieu, C. Measles Encephalitis: Towards New Therapeutics. Viruses 2019, 11, 1017. [Google Scholar] [CrossRef] [Green Version]
- Holzmann, H.; Hengel, H.; Tenbusch, M.; Doerr, H.W. Eradication of measles: Remaining challenges. Med. Microbiol. Immunol. 2016, 205, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Young, M.K.; Nimmo, G.R.; Cripps, A.W.; Jones, M.A. Post-exposure passive immunisation for preventing measles. Cochrane Database Syst. Rev. 2014, Cd010056. [Google Scholar] [CrossRef] [Green Version]
- Zingher, A.; Mortimer, P. Convalescent whole blood, plasma and serum in the prophylaxis of measles: Reproduced from JAMA, 12 April 1926; 1180–1187. Rev. Med. Virol. 2005, 15, 407–421. [Google Scholar] [CrossRef]
- Matysiak-Klose, D.; Santibanez, S.; Schwerdtfeger, C.; Koch, J.; von Bernuth, H.; Hengel, H.; Littmann, M.; Terhardt, M.; Wicker, S.; Mankertz, A.; et al. Post-exposure prophylaxis for measles with immunoglobulins revised recommendations of the standing committee on vaccination in Germany. Vaccine 2018, 36, 7916–7922. [Google Scholar] [CrossRef]
- Grancher, N.; Venard, V.; Kedzierewicz, F.; Ammerlaan, W.; Finance, C.; Muller, C.P.; Le Faou, A. Improved antiviral activity in vitro of ribavirin against measles virus after complexation with cyclodextrins. Antivir. Res. 2004, 62, 135–137. [Google Scholar] [CrossRef]
- Zhang, N.; Chen, H.M.; Sood, R.; Kalicharran, K.; Fattom, A.I.; Naso, R.B.; Barnard, D.L.; Sidwell, R.W.; Hosmane, R.S. In vitro inhibition of the measles virus by novel ring-expanded (‘fat’) nucleoside analogues containing the imidazo[4,5-e]diazepine ring system. Bioorg. Med. Chem. Lett. 2002, 12, 3391–3394. [Google Scholar] [CrossRef]
- Murphy, M.F. In vitro inhibition of subacute sclerosing panencephalitis virus by the antiviral agent ribavirin. J. Infect. Dis. 1978, 138, 249–251. [Google Scholar] [CrossRef] [PubMed]
- Hosoya, M.; Shigeta, S.; Nakamura, K.; De Clercq, E. Inhibitory effect of selected antiviral compounds on measles (SSPE) virus replication in vitro. Antivir. Res. 1989, 12, 87–97. [Google Scholar] [CrossRef]
- Takahashi, T.; Hosoya, M.; Kimura, K.; Ohno, K.; Mori, S.; Takahashi, K.; Shigeta, S. The cooperative effect of interferon-alpha and ribavirin on subacute sclerosing panencephalitis (SSPE) virus infections, in vitro and in vivo. Antivir. Res. 1998, 37, 29–35. [Google Scholar] [CrossRef]
- Krajczyk, A.; Kulinska, K.; Kulinski, T.; Hurst, B.L.; Day, C.W.; Smee, D.F.; Ostrowski, T.; Januszczyk, P.; Zeidler, J. Antivirally active ribavirin analogues—4,5-disubstituted 1,2,3-triazole nucleosides: Biological evaluation against certain respiratory viruses and computational modelling. Antivir. Chem. Chemother. 2014, 23, 161–171. [Google Scholar] [CrossRef]
- Shigeta, S.; Mori, S.; Baba, M.; Ito, M.; Honzumi, K.; Nakamura, K.; Oshitani, H.; Numazaki, Y.; Matsuda, A.; Obara, T.; et al. Antiviral activities of ribavirin, 5-ethynyl-1-beta-D-ribofuranosylimidazole-4-carboxamide, and 6′-(R)-6′-C-methylneplanocin A against several ortho- and paramyxoviruses. Antimicrob. Agents Chemother. 1992, 36, 435–439. [Google Scholar] [CrossRef] [Green Version]
- De Clercq, E.; Montgomery, J.A. Broad-spectrum antiviral activity of the carbocyclic analog of 3-deazaadenosine. Antivir. Res. 1983, 3, 17–24. [Google Scholar] [CrossRef]
- Lo, M.K.; Jordan, R.; Arvey, A.; Sudhamsu, J.; Shrivastava-Ranjan, P.; Hotard, A.L.; Flint, M.; McMullan, L.K.; Siegel, D.; Clarke, M.O.; et al. GS-5734 and its parent nucleoside analog inhibit Filo-, Pneumo-, and Paramyxoviruses. Sci. Rep. 2017, 7, 43395. [Google Scholar] [CrossRef]
- Lo, M.K.; Shrivastava-Ranjan, P.; Chatterjee, P.; Flint, M.; Beadle, J.R.; Valiaeva, N.; Schooley, R.T.; Hostetler, K.Y.; Montgomery, J.M.; Spiropoulou, C. Broad-spectrum in vitro antiviral activity of ODBG-P-RVn: An orally-available, lipid-modified monophosphate prodrug of remdesivir parent nucleoside (GS-441524). bioRxiv 2021. [Google Scholar] [CrossRef]
- Hashimoto, K.; Maeda, H.; Miyazaki, K.; Watanabe, M.; Norito, S.; Maeda, R.; Kume, Y.; Ono, T.; Chishiki, M.; Suyama, K.; et al. Antiviral Effect of Favipiravir (T-705) against Measles and Subacute Sclerosing Panencephalitis Viruses. Jpn. J. Infect. Dis. 2021, 74, 154–156. [Google Scholar] [CrossRef]
- Jochmans, D.; van Nieuwkoop, S.; Smits, S.L.; Neyts, J.; Fouchier, R.A.; van den Hoogen, B.G. Antiviral Activity of Favipiravir (T-705) against a Broad Range of Paramyxoviruses In Vitro and against Human Metapneumovirus in Hamsters. Antimicrob. Agents Chemother. 2016, 60, 4620–4629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, M.K.; Jordan, P.C.; Stevens, S.; Tam, Y.; Deval, J.; Nichol, S.T.; Spiropoulou, C.F. Susceptibility of paramyxoviruses and filoviruses to inhibition by 2′-monofluoro- and 2′-difluoro-4′-azidocytidine analogs. Antivir. Res. 2018, 153, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Barnard, D.L.; Stowell, V.D.; Seley, K.L.; Hegde, V.R.; Das, S.R.; Rajappan, V.P.; Schneller, S.W.; Smee, D.F.; Sidwell, R.W. Inhibition of measles virus replication by 5’-nor carbocyclic adenosine analogues. Antivir. Chem. Chemother. 2001, 12, 241–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGuigan, C.; Hinsinger, K.; Farleigh, L.; Pathirana, R.N.; Bugert, J.J. Novel antiviral activity of l-dideoxy bicyclic nucleoside analogues versus vaccinia and measles viruses in vitro. J. Med. Chem. 2013, 56, 1311–1322. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.J.; Krumm, S.A.; Ndungu, J.M.; Hoffman, V.; Bankamp, B.; Rota, P.A.; Sun, A.; Snyder, J.P.; Plemper, R.K. Target analysis of the experimental measles therapeutic AS-136A. Antimicrob. Agents Chemother. 2009, 53, 3860–3870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ndungu, J.M.; Krumm, S.A.; Yan, D.; Arrendale, R.F.; Reddy, G.P.; Evers, T.; Howard, R.; Natchus, M.G.; Saindane, M.T.; Liotta, D.C.; et al. Non-nucleoside inhibitors of the measles virus RNA-dependent RNA polymerase: Synthesis, structure-activity relationships, and pharmacokinetics. J. Med. Chem. 2012, 55, 4220–4230. [Google Scholar] [CrossRef] [Green Version]
- Krumm, S.A.; Yan, D.; Hovingh, E.S.; Evers, T.J.; Enkirch, T.; Reddy, G.P.; Sun, A.; Saindane, M.T.; Arrendale, R.F.; Painter, G.; et al. An orally available, small-molecule polymerase inhibitor shows efficacy against a lethal morbillivirus infection in a large animal model. Sci. Transl. Med. 2014, 6, 232ra252. [Google Scholar] [CrossRef] [Green Version]
- Cox, R.M.; Sourimant, J.; Govindarajan, M.; Natchus, M.G.; Plemper, R.K. Therapeutic targeting of measles virus polymerase with ERDRP-0519 suppresses all RNA synthesis activity. PLoS Pathog. 2021, 17, e1009371. [Google Scholar] [CrossRef]
- Wittwer, K.; Anderson, D.E.; Pfeffermann, K.; Cox, R.M.; Wolf, J.D.; Santibanez, S.; Mankertz, A.; Plesker, R.; Sticher, Z.M.; Kolkykhalov, A.A.; et al. Small-molecule polymerase inhibitor protects non-human primates from measles and reduces shedding. Nat. Commun. 2021, 12, 5233. [Google Scholar] [CrossRef]
- Cox, R.M.; Sourimant, J.; Toots, M.; Yoon, J.J.; Ikegame, S.; Govindarajan, M.; Watkinson, R.E.; Thibault, P.; Makhsous, N.; Lin, M.J.; et al. Orally efficacious broad-spectrum allosteric inhibitor of paramyxovirus polymerase. Nat. Microbiol. 2020, 5, 1232–1246. [Google Scholar] [CrossRef]
- Richardson, C.D.; Scheid, A.; Choppin, P.W. Specific inhibition of paramyxovirus and myxovirus replication by oligopeptides with amino acid sequences similar to those at the N-termini of the F1 or HA2 viral polypeptides. Virology 1980, 105, 205–222. [Google Scholar] [CrossRef]
- Malvoisin, E.; Wild, F. Effect of drugs which inhibit cholesterol synthesis on syncytia formation in vero cells infected with measles virus. Biochim. Biophys. Acta 1990, 1042, 359–364. [Google Scholar] [CrossRef]
- Ha, M.N.; Delpeut, S.; Noyce, R.S.; Sisson, G.; Black, K.M.; Lin, L.T.; Bilimoria, D.; Plemper, R.K.; Privé, G.G.; Richardson, C.D. Mutations in the Fusion Protein of Measles Virus That Confer Resistance to the Membrane Fusion Inhibitors Carbobenzoxy-d-Phe-l-Phe-Gly and 4-Nitro-2-Phenylacetyl Amino-Benzamide. J. Virol. 2017, 91, e01026-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plemper, R.K.; Doyle, J.; Sun, A.; Prussia, A.; Cheng, L.T.; Rota, P.A.; Liotta, D.C.; Snyder, J.P.; Compans, R.W. Design of a small-molecule entry inhibitor with activity against primary measles virus strains. Antimicrob. Agents Chemother. 2005, 49, 3755–3761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashiguchi, T.; Fukuda, Y.; Matsuoka, R.; Kuroda, D.; Kubota, M.; Shirogane, Y.; Watanabe, S.; Tsumoto, K.; Kohda, D.; Plemper, R.K.; et al. Structures of the prefusion form of measles virus fusion protein in complex with inhibitors. Proc. Natl. Acad. Sci. USA 2018, 115, 2496–2501. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Hashimoto, K.; Abe, Y.; Kodama, E.N.; Nabika, R.; Oishi, S.; Ohara, S.; Sato, M.; Kawasaki, Y.; Fujii, N.; et al. A Novel Peptide Derived from the Fusion Protein Heptad Repeat Inhibits Replication of Subacute Sclerosing Panencephalitis Virus In Vitro and In Vivo. PLoS ONE 2016, 11, e0162823. [Google Scholar] [CrossRef] [Green Version]
- Welsch, J.C.; Talekar, A.; Mathieu, C.; Pessi, A.; Moscona, A.; Horvat, B.; Porotto, M. Fatal measles virus infection prevented by brain-penetrant fusion inhibitors. J. Virol. 2013, 87, 13785–13794. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, C.; Huey, D.; Jurgens, E.; Welsch, J.C.; DeVito, I.; Talekar, A.; Horvat, B.; Niewiesk, S.; Moscona, A.; Porotto, M. Prevention of measles virus infection by intranasal delivery of fusion inhibitor peptides. J. Virol. 2015, 89, 1143–1155. [Google Scholar] [CrossRef] [Green Version]
- Figueira, T.N.; Palermo, L.M.; Veiga, A.S.; Huey, D.; Alabi, C.A.; Santos, N.C.; Welsch, J.C.; Mathieu, C.; Horvat, B.; Niewiesk, S.; et al. In Vivo Efficacy of Measles Virus Fusion Protein-Derived Peptides Is Modulated by the Properties of Self-Assembly and Membrane Residence. J. Virol. 2017, 91, e01554-16. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, C.; Ferren, M.; Jurgens, E.; Dumont, C.; Rybkina, K.; Harder, O.; Stelitano, D.; Madeddu, S.; Sanna, G.; Schwartz, D.; et al. Measles Virus Bearing Measles Inclusion Body Encephalitis-Derived Fusion Protein Is Pathogenic after Infection via the Respiratory Route. J. Virol. 2019, 93, e01862-18. [Google Scholar] [CrossRef] [Green Version]
- Bovier, F.T.; Rybkina, K.; Biswas, S.; Harder, O.; Marcink, T.C.; Niewiesk, S.; Moscona, A.; Alabi, C.A.; Porotto, M. Inhibition of Measles Viral Fusion Is Enhanced by Targeting Multiple Domains of the Fusion Protein. ACS Nano 2021, 15, 12794–12803. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.; Prussia, A.; Zhan, W.; Murray, E.E.; Doyle, J.; Cheng, L.T.; Yoon, J.J.; Radchenko, E.V.; Palyulin, V.A.; Compans, R.W.; et al. Nonpeptide inhibitors of measles virus entry. J. Med. Chem. 2006, 49, 5080–5092. [Google Scholar] [CrossRef] [PubMed]
- White, L.K.; Yoon, J.J.; Lee, J.K.; Sun, A.; Du, Y.; Fu, H.; Snyder, J.P.; Plemper, R.K. Nonnucleoside inhibitor of measles virus RNA-dependent RNA polymerase complex activity. Antimicrob. Agents Chemother. 2007, 51, 2293–2303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ader, N.; Brindley, M.; Avila, M.; Örvell, C.; Horvat, B.; Hiltensperger, G.; Schneider-Schaulies, J.; Vandevelde, M.; Zurbriggen, A.; Plemper, R.K.; et al. Mechanism for active membrane fusion triggering by morbillivirus attachment protein. J. Virol. 2013, 87, 314–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plemper, R.K.; Erlandson, K.J.; Lakdawala, A.S.; Sun, A.; Prussia, A.; Boonsombat, J.; Aki-Sener, E.; Yalcin, I.; Yildiz, I.; Temiz-Arpaci, O.; et al. A target site for template-based design of measles virus entry inhibitors. Proc. Natl. Acad. Sci. USA 2004, 101, 5628–5633. [Google Scholar] [CrossRef] [Green Version]
- Zinke, M.; Kendl, S.; Singethan, K.; Fehrholz, M.; Reuter, D.; Rennick, L.; Herold, M.J.; Schneider-Schaulies, J. Clearance of measles virus from persistently infected cells by short hairpin RNA. J. Virol. 2009, 83, 9423–9431. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, N.; Gall, F.M.; Mathieu, C.; Hierweger, M.M.; Brügger, M.; Alves, M.P.; Vesin, J.; Banfi, D.; Kalbermatter, D.; Horvat, B.; et al. Highly Potent Host-Specific Small-Molecule Inhibitor of Paramyxovirus and Pneumovirus Replication with High Resistance Barrier. mBio 2021, 12, e0262121. [Google Scholar] [CrossRef]
- Moore, T.W.; Sana, K.; Yan, D.; Krumm, S.A.; Thepchatri, P.; Snyder, J.P.; Marengo, J.; Arrendale, R.F.; Prussia, A.J.; Natchus, M.G.; et al. Synthesis and Metabolic Studies of Host Directed Inhibitors for Anti Viral Therapy. ACS Med. Chem. Lett. 2013, 4, 762–767. [Google Scholar] [CrossRef]
- Wachsman, M.B.; Ramirez, J.A.; Galagovsky, L.R.; Coto, C.E. Antiviral activity of brassinosteroids derivatives against measles virus in cell cultures. Antivir. Chem. Chemother. 2002, 13, 61–66. [Google Scholar] [CrossRef]
- Morán-Santibañez, K.; Peña-Hernández, M.A.; Cruz-Suárez, L.E.; Ricque-Marie, D.; Skouta, R.; Vasquez, A.H.; Rodríguez-Padilla, C.; Trejo-Avila, L.M. Virucidal and Synergistic Activity of Polyphenol-Rich Extracts of Seaweeds against Measles Virus. Viruses 2018, 10, 465. [Google Scholar] [CrossRef] [Green Version]
- Olila, D.; Olwa, O.; Opuda-Asibo, J. Screening extracts of Zanthoxylum chalybeum and Warburgia ugandensis for activity against measles virus (Swartz and Edmonston strains) in vitro. Afr. Health Sci. 2002, 2, 2–10. [Google Scholar] [PubMed]
- Trejo-Avila, L.M.; Morales-Martínez, M.E.; Ricque-Marie, D.; Cruz-Suarez, L.E.; Zapata-Benavides, P.; Morán-Santibañez, K.; Rodríguez-Padilla, C. In vitro anti-canine distemper virus activity of fucoidan extracted from the brown alga Cladosiphon okamuranus. Virus Dis. 2014, 25, 474–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nwodo, U.U.; Ngene, A.A.; Iroegbu, C.U.; Onyedikachi, O.A.; Chigor, V.N.; Okoh, A.I. In vivo evaluation of the antiviral activity of Cajanus cajan on measles virus. Arch. Virol. 2011, 156, 1551–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedows, E.; Hatfield, G.M. An investigation of the antiviral activity of Podophyllum peltatum. J. Nat. Prod. 1982, 45, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, M.; Ochiai, H.; Nagasaka, K.; Neki, M.; Xu, H.; Kadota, S.; Sutardjo, S.; Matsumoto, T.; Namba, T.; Shiraki, K. Antiviral traditional medicines against herpes simplex virus (HSV-1), poliovirus, and measles virus in vitro and their therapeutic efficacies for HSV-1 infection in mice. Antivir. Res. 1993, 22, 175–188. [Google Scholar] [CrossRef]
- Parker, M.E.; Chabot, S.; Ward, B.J.; Johns, T. Traditional dietary additives of the Maasai are antiviral against the measles virus. J. Ethnopharmacol. 2007, 114, 146–152. [Google Scholar] [CrossRef]
- Sanekata, T.; Fukuda, T.; Miura, T.; Morino, H.; Lee, C.; Maeda, K.; Araki, K.; Otake, T.; Kawahata, T.; Shibata, T. Evaluation of the antiviral activity of chlorine dioxide and sodium hypochlorite against feline calicivirus, human influenza virus, measles virus, canine distemper virus, human herpesvirus, human adenovirus, canine adenovirus and canine parvovirus. Biocontrol Sci. 2010, 15, 45–49. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, F.; Imatani, Y.; Nagaki, D.; Nakagawa, A.; Omura, S. Selective antiviral activity of the antibiotic 2′-amino-2′-deoxyribofuranosyl adenine. J. Antibiot. 1981, 34, 313–316. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Zhao, Z.; Zhou, D.; Chen, Y.; Hong, W.; Cao, L.; Yang, J.; Zhang, Y.; Shi, W.; Cao, Z.; et al. Virucidal activity of a scorpion venom peptide variant mucroporin-M1 against measles, SARS-CoV and influenza H5N1 viruses. Peptides 2011, 32, 1518–1525. [Google Scholar] [CrossRef]
- Miller, F.A.; Dixon, G.J.; Arnett, G.; Dice, J.R.; Rightsel, W.A.; Schabel, F.M., Jr.; McLean, I.W., Jr. Antiviral activity of carbobenzosy di- and tripeptides on measles virus. Appl. Microbiol. 1968, 16, 1489–1496. [Google Scholar] [CrossRef]
- Sleeman, K.; Stein, D.A.; Tamin, A.; Reddish, M.; Iversen, P.L.; Rota, P.A. Inhibition of measles virus infections in cell cultures by peptide-conjugated morpholino oligomers. Virus Res. 2009, 140, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Verini, M.A.; Fioretti, A.; Casazza, A.M.; Sanfilippo, A.; Palamidessi, G.; Ghione, M. Antiviral activity of a pyrazino-pyrazine derivative. Chemotherapy 1975, 21, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Sugita, K.; Arita, H.; Kawanami, J.; Sato, K. Studies on antiviral glycosides, 4. Inhibition of the multiplication of paramyxoviruses by phenyl-6-chloro-6-deoxy-beta-D-glucopyranoside. J. Gen. Virol. 1979, 45, 249–251. [Google Scholar] [CrossRef]
- Fletcher, N.F.; Meredith, L.W.; Tidswell, E.L.; Bryden, S.R.; Gonçalves-Carneiro, D.; Chaudhry, Y.; Shannon-Lowe, C.; Folan, M.A.; Lefteri, D.A.; Pingen, M.; et al. A novel antiviral formulation inhibits a range of enveloped viruses. J. Gen. Virol. 2020, 101, 1090–1102. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.W.; Weinstein, L. Antiviral activity of isoprinosine in vitro and in vivo. Am. J. Med. Sci. 1973, 265, 143–146. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E.; Neyts, J. Antiviral agents acting as DNA or RNA chain terminators. In Handbook of Experimental Pharmacology; Springer: Berlin/Heideberg, Germany, 2009; pp. 53–84. [Google Scholar] [CrossRef]
- Eltahla, A.A.; Luciani, F.; White, P.A.; Lloyd, A.R.; Bull, R.A. Inhibitors of the Hepatitis C Virus Polymerase; Mode of Action and Resistance. Viruses 2015, 7, 5206–5224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidwell, R.W.; Huffman, J.H.; Khare, G.P.; Allen, L.B.; Witkowski, J.T.; Robins, R.K. Broad-spectrum antiviral activity of Virazole: 1-beta-D-ribofuranosyl-1,2,4-triazole-3-carboxamide. Science 1972, 177, 705–706. [Google Scholar] [CrossRef]
- Geraghty, R.J.; Aliota, M.T.; Bonnac, L.F. Broad-Spectrum Antiviral Strategies and Nucleoside Analogues. Viruses 2021, 13, 667. [Google Scholar] [CrossRef]
- Wright, P.J.; Crameri, G.; Eaton, B.T. RNA synthesis during infection by Hendra virus: An examination by quantitative real-time PCR of RNA accumulation, the effect of ribavirin and the attenuation of transcription. Arch. Virol. 2005, 150, 521–532. [Google Scholar] [CrossRef]
- Crotty, S.; Andino, R. Implications of high RNA virus mutation rates: Lethal mutagenesis and the antiviral drug ribavirin. Microbes Infect. 2002, 4, 1301–1307. [Google Scholar] [CrossRef]
- Agostini, M.L.; Andres, E.L.; Sims, A.C.; Graham, R.L.; Sheahan, T.P.; Lu, X.; Smith, E.C.; Case, J.B.; Feng, J.Y.; Jordan, R.; et al. Coronavirus Susceptibility to the Antiviral Remdesivir (GS-5734) Is Mediated by the Viral Polymerase and the Proofreading Exoribonuclease. mBio 2018, 9, e00221-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warren, T.K.; Jordan, R.; Lo, M.K.; Ray, A.S.; Mackman, R.L.; Soloveva, V.; Siegel, D.; Perron, M.; Bannister, R.; Hui, H.C.; et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 2016, 531, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Gilead. Press Releases: U. S. Food and Drug Administration Approves Gilead’s Antiviral Veklury (Remdesivir) for Treatment of COVID-19. Available online: https://www.gilead.com/news-and-press/press-room/press-releases/2020/10/us-food-and-drug-administration-approves-gileads-antiviral-veklury-remdesivir-for-treatment-of-covid19 (accessed on 20 February 2022).
- Kumar, R.; Mishra, S.; Shreya; Maurya, S.K. Recent advances in the discovery of potent RNA-dependent RNA-polymerase (RdRp) inhibitors targeting viruses. RSC Med. Chem. 2021, 12, 306–320. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.J.; Chawla, D.; Paal, T.; Ndungu, M.; Du, Y.; Kurtkaya, S.; Sun, A.; Snyder, J.P.; Plemper, R.K. High-throughput screening-based identification of paramyxovirus inhibitors. J. Biomol. Screen. 2008, 13, 591–608. [Google Scholar] [CrossRef] [Green Version]
- De Vries, R.D.; Yüksel, S.; Osterhaus, A.D.; de Swart, R.L. Specific CD8(+) T-lymphocytes control dissemination of measles virus. Eur. J. Immunol. 2010, 40, 388–395. [Google Scholar] [CrossRef]
- Davis, M.E.; Wang, M.K.; Rennick, L.J.; Full, F.; Gableske, S.; Mesman, A.W.; Gringhuis, S.I.; Geijtenbeek, T.B.; Duprex, W.P.; Gack, M.U. Antagonism of the phosphatase PP1 by the measles virus V protein is required for innate immune escape of MDA5. Cell Host Microbe 2014, 16, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Rennick, L.J.; de Vries, R.D.; Carsillo, T.J.; Lemon, K.; van Amerongen, G.; Ludlow, M.; Nguyen, D.T.; Yüksel, S.; Verburgh, R.J.; Haddock, P.; et al. Live-attenuated measles virus vaccine targets dendritic cells and macrophages in muscle of nonhuman primates. J. Virol. 2015, 89, 2192–2200. [Google Scholar] [CrossRef] [Green Version]
- Laksono, B.M.; de Vries, R.D.; Verburgh, R.J.; Visser, E.G.; de Jong, A.; Fraaij, P.L.A.; Ruijs, W.L.M.; Nieuwenhuijse, D.F.; van den Ham, H.-J.; Koopmans, M.P.G.; et al. Studies into the mechanism of measles-associated immune suppression during a measles outbreak in the Netherlands. Nat. Commun. 2018, 9, 4944. [Google Scholar] [CrossRef]
- Young, M.K. The indications and safety of polyvalent immunoglobulin for post-exposure prophylaxis of hepatitis A, rubella and measles. Hum. Vaccines Immunother. 2019, 15, 2060–2065. [Google Scholar] [CrossRef]
- Janeway, C.A. Use of Concentrated Human Serum Gamma-Globulin in the Prevention and Attenuation of Measles. Bull. N. Y. Acad. Med. 1945, 21, 202–222. [Google Scholar]
- De Vries, R.D.; Schmitz, K.S.; Bovier, F.T.; Predella, C.; Khao, J.; Noack, D.; Haagmans, B.L.; Herfst, S.; Stearns, K.N.; Drew-Bear, J.; et al. Intranasal fusion inhibitory lipopeptide prevents direct-contact SARS-CoV-2 transmission in ferrets. Science 2021, 371, 1379–1382. [Google Scholar] [CrossRef] [PubMed]
- Wild, C.T.; Shugars, D.C.; Greenwell, T.K.; McDanal, C.B.; Matthews, T.J. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. USA 1994, 91, 9770–9774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalezari, J.P. Clinical safety and efficacy of enfuvirtide (T-20), a new fusion inhibitor. A review of the presentation at the satellite symposium “New hope: Advancing care in HIV infection” at the 15th annual Association of Nurses in AIDS Care conference, November 2002. AIDS Read. 2003, 13, S9–S13. [Google Scholar] [PubMed]
- Greenberg, M.L.; Cammack, N. Resistance to enfuvirtide, the first HIV fusion inhibitor. J. Antimicrob. Chemother. 2004, 54, 333–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, J.; Prussia, A.; White, L.K.; Sun, A.; Liotta, D.C.; Snyder, J.P.; Compans, R.W.; Plemper, R.K. Two domains that control prefusion stability and transport competence of the measles virus fusion protein. J. Virol. 2006, 80, 1524–1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingallinella, P.; Bianchi, E.; Ladwa, N.A.; Wang, Y.J.; Hrin, R.; Veneziano, M.; Bonelli, F.; Ketas, T.J.; Moore, J.P.; Miller, M.D.; et al. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc. Natl. Acad. Sci. USA 2009, 106, 5801–5806. [Google Scholar] [CrossRef] [Green Version]
- Porotto, M.; Rockx, B.; Yokoyama, C.C.; Talekar, A.; Devito, I.; Palermo, L.M.; Liu, J.; Cortese, R.; Lu, M.; Feldmann, H.; et al. Inhibition of Nipah virus infection in vivo: Targeting an early stage of paramyxovirus fusion activation during viral entry. PLoS Pathog. 2010, 6, e1001168. [Google Scholar] [CrossRef]
- Von Messling, V.; Springfeld, C.; Devaux, P.; Cattaneo, R. A ferret model of canine distemper virus virulence and immunosuppression. J. Virol. 2003, 77, 12579–12591. [Google Scholar] [CrossRef] [Green Version]
- Niewiesk, S. Current animal models: Cotton rat animal model. Curr. Top. Microbiol. Immunol. 2009, 330, 89–110. [Google Scholar] [CrossRef]
- Ohuchi, R.; Ohuchi, M.; Mifune, K. Slow development of measles virus (Edmonston strain) infection in the brain of nude mice. Microbiol. Immunol. 1984, 28, 757–764. [Google Scholar] [CrossRef] [Green Version]
- Honda, Y.; Hosoya, M.; Ishii, T.; Shigeta, S.; Suzuki, H. Effect of ribavirin on subacute sclerosing panencephalitis virus infections in hamsters. Antimicrob. Agents Chemother. 1994, 38, 653–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, T.; Hosoya, M.; Mori, S.; Shigeta, S.; Suzuki, H. Effective ribavirin concentration in hamster brains for antiviral chemotherapy for subacute sclerosing panencephalitis. Antimicrob. Agents Chemother. 1996, 40, 241–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosoya, M.; Shigeta, S.; Mori, S.; Tomoda, A.; Shiraishi, S.; Miike, T.; Suzuki, H. High-dose intravenous ribavirin therapy for subacute sclerosing panencephalitis. Antimicrob. Agents Chemother. 2001, 45, 943–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosoya, M.; Mori, S.; Tomoda, A.; Mori, K.; Sawaishi, Y.; Kimura, H.; Shigeta, S.; Suzuki, H. Pharmacokinetics and effects of ribavirin following intraventricular administration for treatment of subacute sclerosing panencephalitis. Antimicrob. Agents Chemother. 2004, 48, 4631–4635. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antiviral Class | Examples Investigated for MV In Vitro |
|---|---|
| Nucleoside analogs | Ribavirin and ribavirin-derivatives and -analogs [33,34,35,36,37,38,39,40] Ring-expanded purine nucleosides [34] Remdesivir [41,42] Favipiravir (T-705) [43,44] Derivatives of R1479 [45] Pyrazofurin [36] 3-Deazaguanine and its carbocyclic analog [36,38,40] 6-Azauridine [36] 5’-nor carbocyclic adenosine analogues [46] L-dideoxy bicyclic nucleoside analogs [47] |
| Non-nucleoside polymerase inhibitors | AS136a [48,49] ERDRP-0519 [49,50,51,52] GHP-88309 [53] |
| Fusion and entry inhibitors | Peptide inhibitors Z-D- Phe-L-Phe-L-Gly (FIP) [54,55,56,57,58] HRC-based peptides: M1, M2, M3, M4, M1EK, M2EK, M3EK, M4EK [59] HRC-lipopeptides [60,61,62,63,64] FIP-lipopeptides [64] FIP-HRC-lipopeptides [64] Nonpeptidic small molecules [65] 16677 [66] 3G [63,67] AS-48 [56,57,58,65,66] OX-1-(variants) [57,68] AM-2 [68] AM-4 [57] shRNA [69] |
| Fusion and entry inhibitors | |
| Host-directed compounds | ZHAWOC9045 and ZHAWOC21026 [70] EMXV-001299 and EMXV-1400 [71] Benzimidazole (JMN3-003) [71] |
| Plant extracts | Brassinosteroids [72] Polyphenol-rich extracts from seaweed [73] Extracts of Zanthoxylum chalybeum [74] Fucoidan (brown alga) [75] Extracts from cajanus cajan [76] Extracts from podophyllum peltatum [77] Hot Water extracts [78] Olinia rochetiana (Olkirenyi) extracts [79] Warburgia ugandensis (Osokonoi) extracts [74,79] |
| Disinfectant | Chlorine dioxide and sodium hypochloride [80] |
| Anti-mycoplasmal and anti-microbial peptides | 2´-Amino-2´-Deoxyribofuranosyl Adenine (2-AA) [81] Mucroporin (optimized/attenuated) [82] |
| Interferons | IFNα [37] |
| Others | Inhibitors of cholesterol synthesis (W-7, cerulenin, mevinolin, miconazole, ketoconazole) [55] Carbobenzoxy-(di-or tri-) peptides [83] PPMO (anti-mRNA) [84] Pyrazino-Pyrazine derivate [85] PCG [86] ViroSAL (emulsion of short-chain caprylic acid) [87] Isoprinosine (inosine derivate) [88] |
| Assay | Description | Cells | Virus Strains |
|---|---|---|---|
| Virus-yield reduction assay | Determination of MV neutralization by a (serially diluted) compound. Compound potency is evaluated by titration of supernatants generated during incubation of MV in the presence of the antiviral compound. Titration can be performed as plaque assay, end-point titration (TCID50) or HA assay. | Vero [55,65,66,68,72,77,88] Vero E6 [82] CV-1 [46] Vero-humanSLAM [44,48,49,50,52,53,57,65,66,87,99] LLCMK [86] HEL-R66 [81] CK [85] U937 [79] human PBMC [52] | MV Edmonston(-derived) Edmonston [46,65,81,82,85] rMV-Edm [48,50,66] rMV-Edm-GFP [44,68] MV wild-type(-derived) MV/Brazil/001/991 [72] MVi/Alaska.USA/16.00 [48,49,50,53,99] MVi/Ibadan.NIE/97/1 [48,50] MVi-Amsterdam.NET/49.97 [48] MVi/Maryland.USA/77 [50] MVi/Illinois.USA/46.02 [50] MVi/NewJersey.USA/94/1 [50,52] MVi/Illinois.USA/50.99 [50] MVi/Kansas.USA/43.00 [57,65] MV Hallé strain [55] Chicago-1 [46] culture adapted Chicago 1 [79] MV-Ibd, MV-JM77, MV-NJ, MV-III 99, MV-Vic/Aus, MV-Amster.NET, MV Gresik, MV-Alaska [66] rMV-IC323-eGFP [87] Undefined MV [53,77,86,88] |
| Neutralization assay | Determination of (complete) neutralization by a (serially diluted) compound. Alternatively: Fixed concentration of compound but serially diluted concentration of virus. Compound potency is evaluated by readout of CPE/syncytia-formation-inhibition. Inhibition can be visually (microscopically) evaluated, potentially supported by crystal violet staining or by staining of the monolayer with neutral red and evaluation of optical density (neutral red inhibition assay). | Vero [36,37,40,59,65,68,73,74,77,88] Vero E6 [38] CV-1 [34,46] Vero-humanSLAM [48,57,59,65,80,99] CHO/SLAM [84] Hep-2 [76,83] B95a [47] BSC-1 [47] CK [85] | MV Edmonston(-derived) Edmonston [46,47,59,65,73,80,83,84,85] rMV-Edm [48] rMV-Edm-GFP [68] Schwarz [74] MV wild-type(-derived) MVi/Kansas.USA/43.00 [57,65] MVi/Alaska.USA/16.00 [48,99] MVi-Amsterdam.NET/49.97 [48] MVi/Ibadan.NIE/97/1 [48] CC [38] WTFb [47] Berkeley/83, Ibadan/97, Chicago1/89 [84] TN1994, Halonen, Bil, X-1108, SA, CC, Chicago-1 [46] Suguyama [36] Hep-2 adapted attenuated MeV [76] SSPE viruses Yamagata-1 [36,37,59] Niigata-1 [36] Kitaken-1 [36] Undefined MV [34,40,77,88] |
| Antigen reduction assay | Determination of (complete) neutralization by a (serially diluted) compound. Compound potency is evaluated by staining of the monolayer with an anti-MV antibody and evaluation of MV antigen expression. | Vero [33,35] HeLa [41] HEL-R66 [81] | MV Edmonston(-derived) Edmonston [33,35,41,81] Adapted MV CAM/RB [33] SSPE viruses Hallé [35] Mantooth [35] McClellan [35] |
| Reporter assay | Determination of (complete) neutralization by a (serially diluted) compound. Compound potency is evaluated by expression of a reporter protein, i.e., fluorescent protein or luciferase. Quantification of the reporter expression indicates potency of the compound. | Vero [68] VeroE6 [42,53] Vero-humanSLAM [44,51,53,60] Vero-dogSLAM [70] CHO/SLAM [56] CHO/CD46 [56] CHO/PVRL4 [56] HeLa [41] NCI-H358 [42,45] HSAEC1-KT [42] NT2 [69] HEK293T [69] | MV Edmonston-derived rMV-Edm-(E)GFP [41,42,44,45,56,68] rMV-Moraten-Luciferase [70] rMV-NanolucPEST [51,53] MV wild-type-derived rMV-IC323-eGFP [56,60] rMV-IC323-Luciferase [70] Adapted MV-derived rMVHcRed-CAMH [69] rMV-EGFP-CAMH [69] |
| Plaque assay | (Serially diluted) compounds added to overlay medium. After fixation, plaques are visualized with crystal violet, neutral red or fluorescently (reporter virus or immunostaining) and used to evaluate compound potency. | Vero [35,39,43,59,63,74,78,79] Vero E6 [82] Vero-humanSLAM [43,60,61,63] Hep-2 [83] HEL-R66 [81] | MV Edmonston(-derived) Edmonston [39,43,74,79,81,82,83] MV wild-type-derived Tanabe strain [78] Sugiyama [39] Toyoshima [39] rMV-IC323-eGFP [63] MV G954 [60,61] SSPE viruses Yamagata-1 [39,43,59] Hallé [35] |
| (Chemiluminescent) complementation-based assay | Cells transiently transfected with a MV receptor (and often one subunit of a reporter) are incubated with cells co-expressing MV glycoproteins H and F (and the other reporter subunit). The fusion process is evaluated in presence of compounds by read-out of reporter expression (i.e., chemiluminescence) or by microscopical analysis of syncytia formation. | Vero [56,67,68,99] Vero-humanSLAM [56,57,67] Vero-PVRL4 [56] HEK293 [56] HEK293T [61,63,64] HEK293T/SLAM [60,61,62,63,64] HEK293T/nectin-4 [62,63,64] | MV Edmonston-derived MV-Edm H, F [56,67,68] MV wild-type-derived MV-Kansas H/F [57] MV-G954 H/F [61] MV-IC323 H/F [60,62,63,64] Undefined MV-derived MV H/F [99] |
| Replicon assay | Evaluates compounds for their effect on the polymerase unit. In the assay a minigenome composed of MV-N, -P and -L and a reporter are used instead of live virus. The reporter can be CAT, a fluorescent or a luciferase protein. Quantification of the reporter evaluates the potency of the antiviral compound. | BHK/sr/T7 [84] BHK-T [48] BSR T7/5 [51,53,66,70] HEK293T [69] | MV Edmonston-derived minigenomes [48,51,53,66,69,70,84] |
| RT-PCR | Determination of neutralization/inhibition by a (serially diluted) compound. Compound potency is evaluated by detecting MV-genomes by RT-PCR. | Vero [48,73] Vero-humanSLAM [51,53] | MV Edmonston(-derived) Edmonston [73] rMV-Edm [48] MV wild-type-derived rMV-Anc (from MVi/Alaska.USA/16.00) [51,53] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmitz, K.S.; Lange, M.V.; Gommers, L.; Handrejk, K.; Porter, D.P.; Alabi, C.A.; Moscona, A.; Porotto, M.; de Vries, R.D.; de Swart, R.L. Repurposing an In Vitro Measles Virus Dissemination Assay for Screening of Antiviral Compounds. Viruses 2022, 14, 1186. https://doi.org/10.3390/v14061186
Schmitz KS, Lange MV, Gommers L, Handrejk K, Porter DP, Alabi CA, Moscona A, Porotto M, de Vries RD, de Swart RL. Repurposing an In Vitro Measles Virus Dissemination Assay for Screening of Antiviral Compounds. Viruses. 2022; 14(6):1186. https://doi.org/10.3390/v14061186
Chicago/Turabian StyleSchmitz, Katharina S., Mona V. Lange, Lennert Gommers, Kim Handrejk, Danielle P. Porter, Christopher A. Alabi, Anne Moscona, Matteo Porotto, Rory D. de Vries, and Rik L. de Swart. 2022. "Repurposing an In Vitro Measles Virus Dissemination Assay for Screening of Antiviral Compounds" Viruses 14, no. 6: 1186. https://doi.org/10.3390/v14061186
APA StyleSchmitz, K. S., Lange, M. V., Gommers, L., Handrejk, K., Porter, D. P., Alabi, C. A., Moscona, A., Porotto, M., de Vries, R. D., & de Swart, R. L. (2022). Repurposing an In Vitro Measles Virus Dissemination Assay for Screening of Antiviral Compounds. Viruses, 14(6), 1186. https://doi.org/10.3390/v14061186