
Structural Plasticity and Immune Evasion of SARS-CoV-2 Spike Variants


Abstract
:1. Introduction
1.1. The Rise of SARS-CoV-2
1.2. The Critical Player: The Spike Glycoprotein
1.3. The Emergence of Different SARS-CoV-2 Spike Variants
2. Virus Transmissibility and Immune Evasion of SARS-CoV-2 VOCs
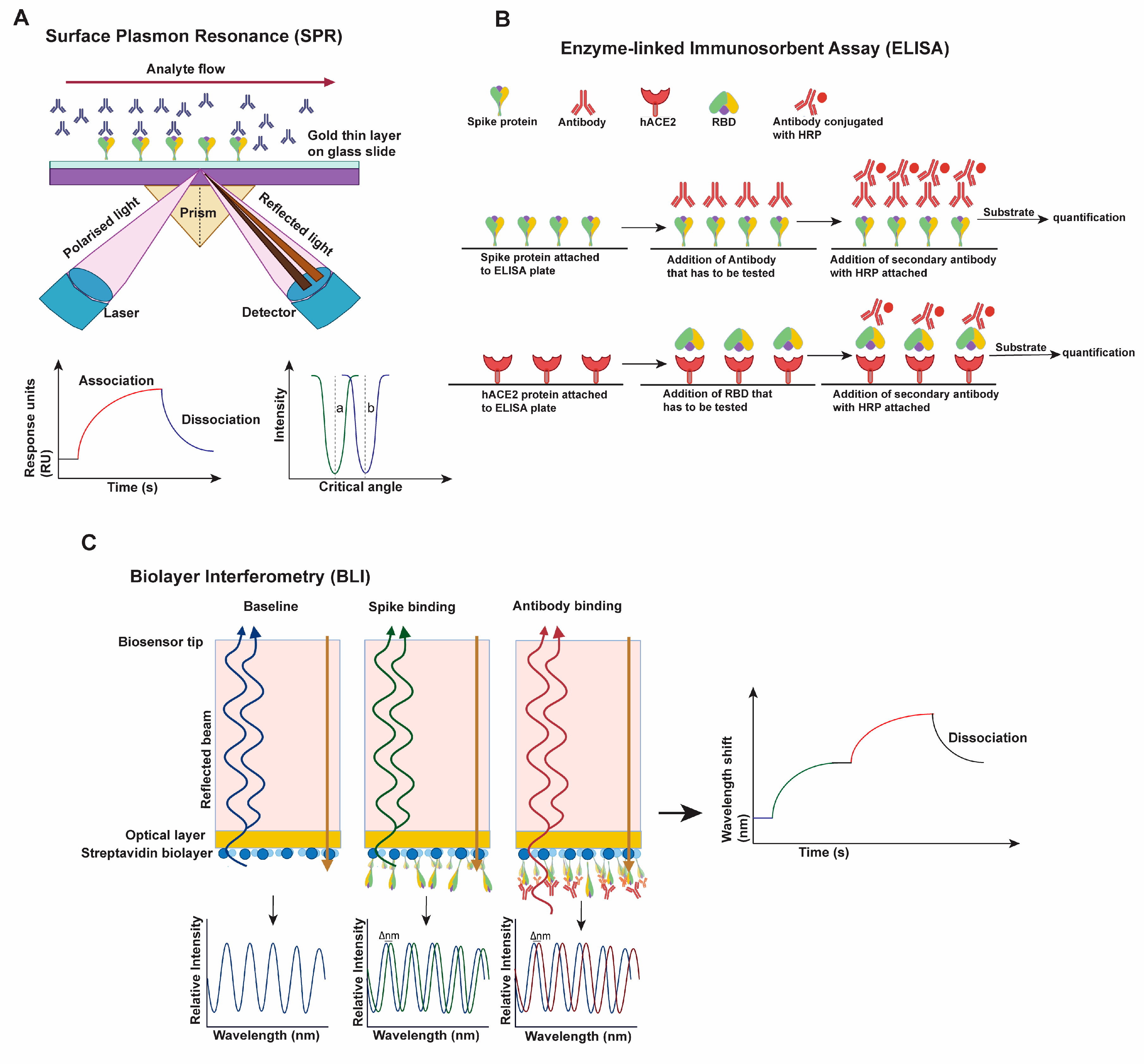
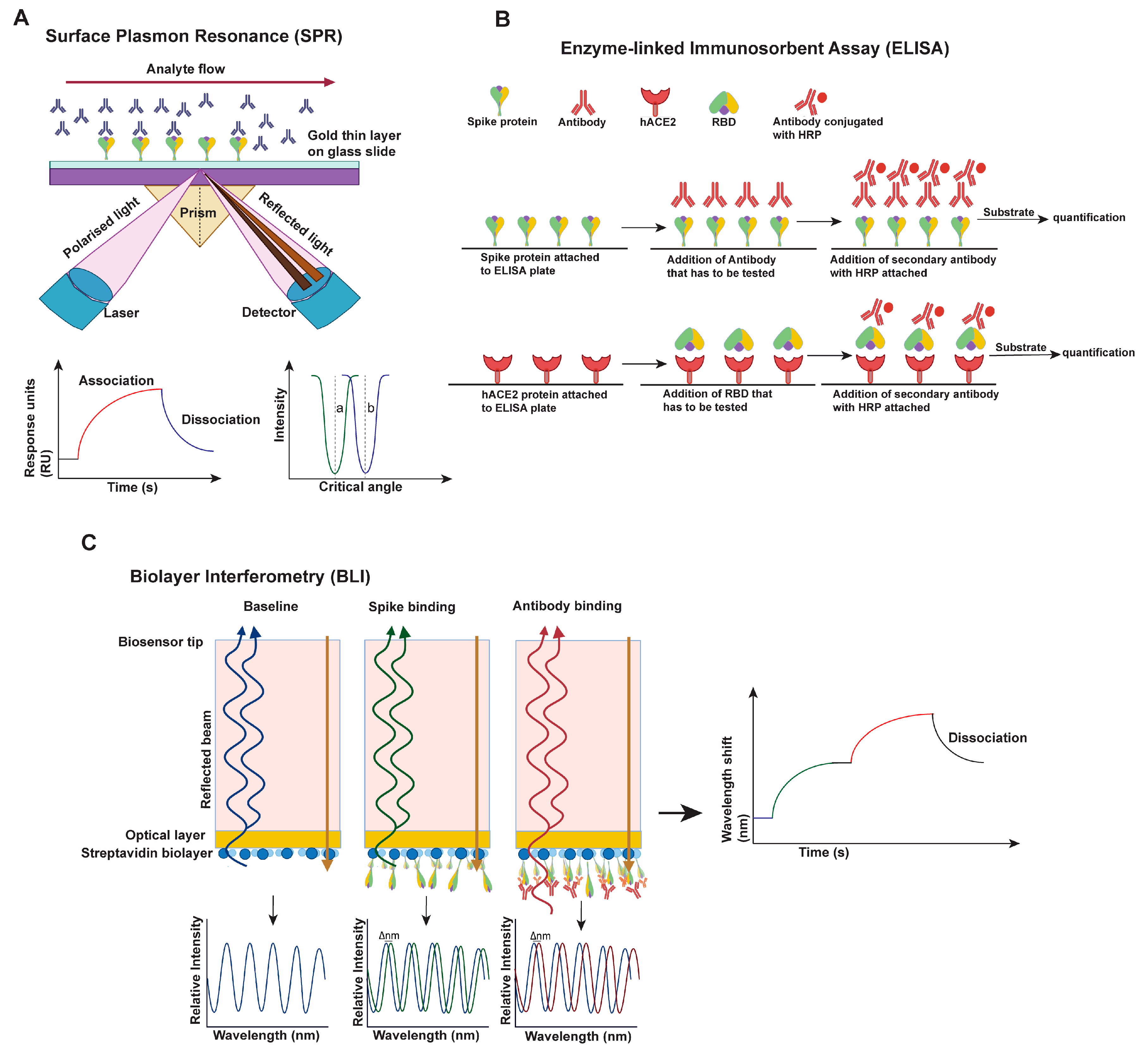
2.1. Approaches Used for Characterizing Virus Transmissibility and Immune Evasion
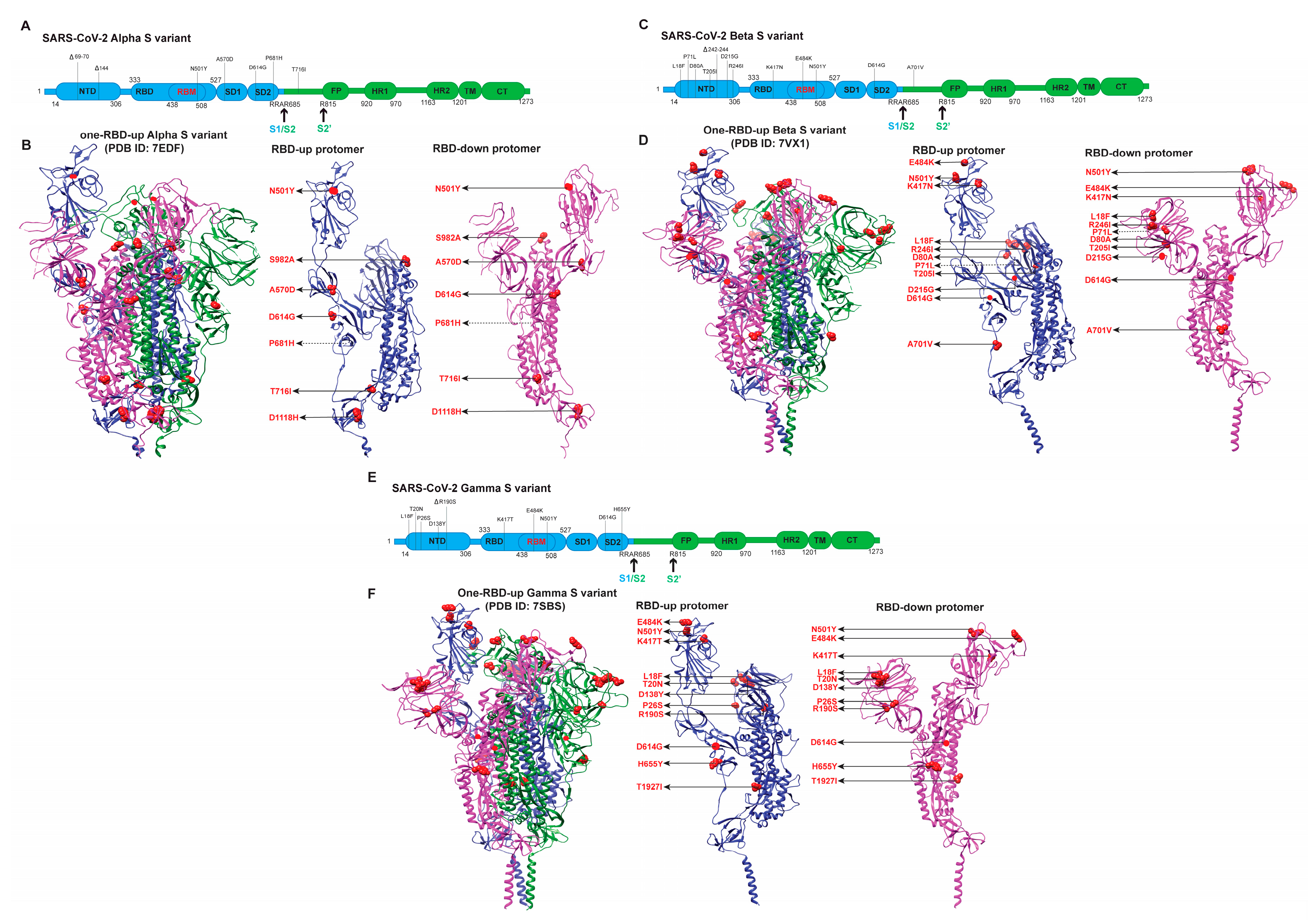
2.2. Early Variants (Alpha, Beta, Gamma)
2.3. Delta/B.1.617.2 Variant
2.4. Omicron/B.1.1.529 Variant
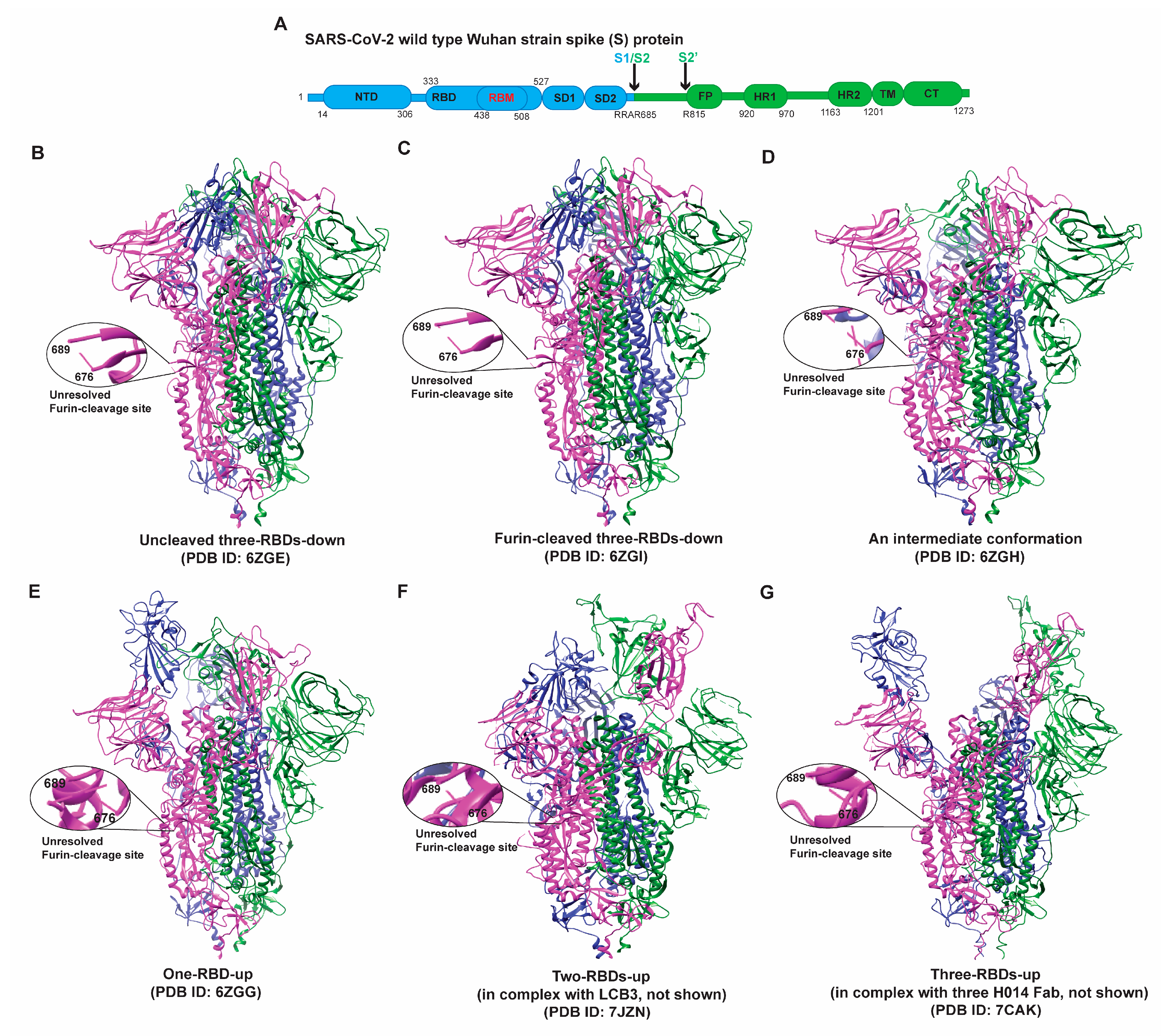
3. Structural Plasticity/Flexibility
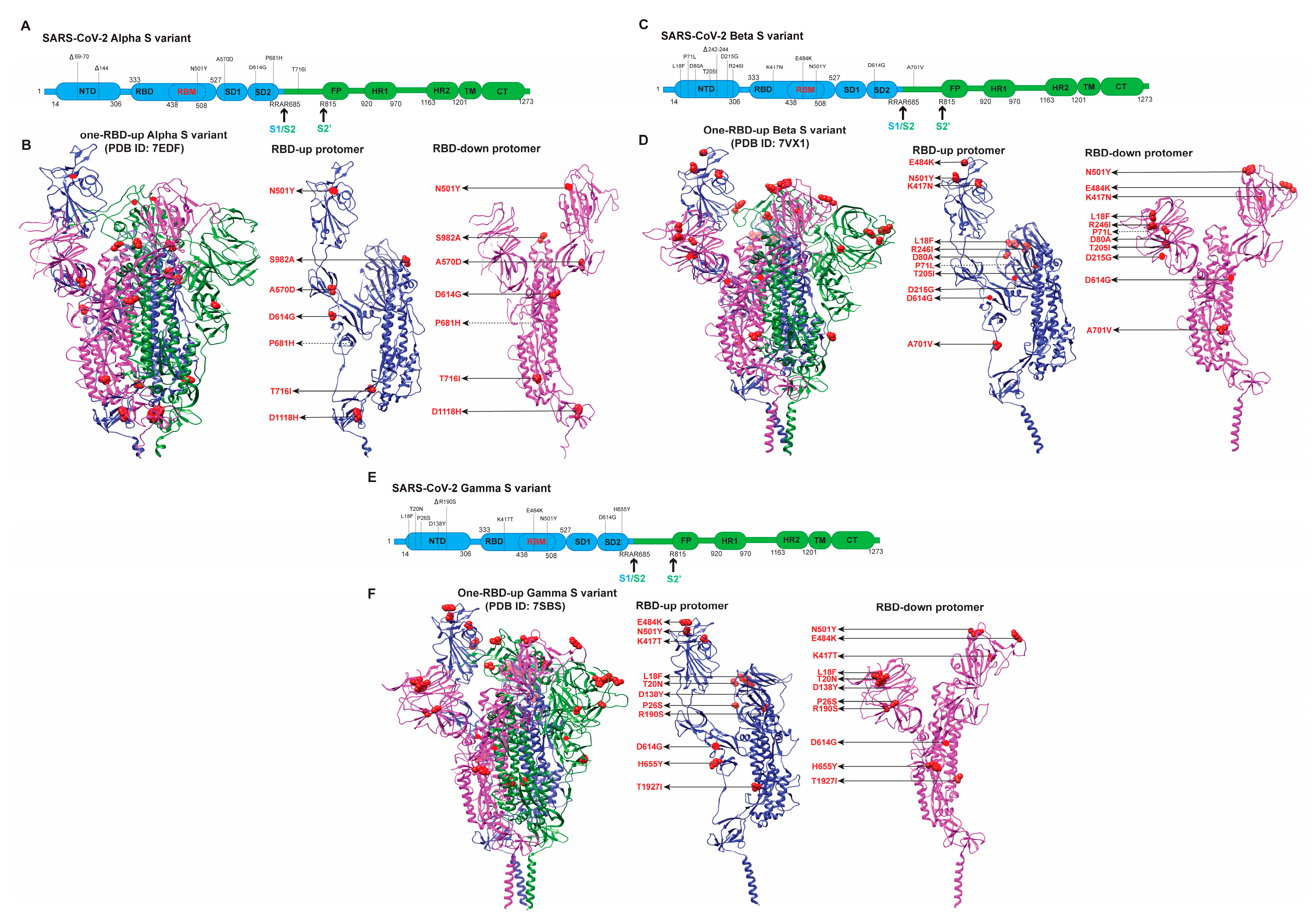
3.1. Early Variants (Alpha, Beta, Gamma)
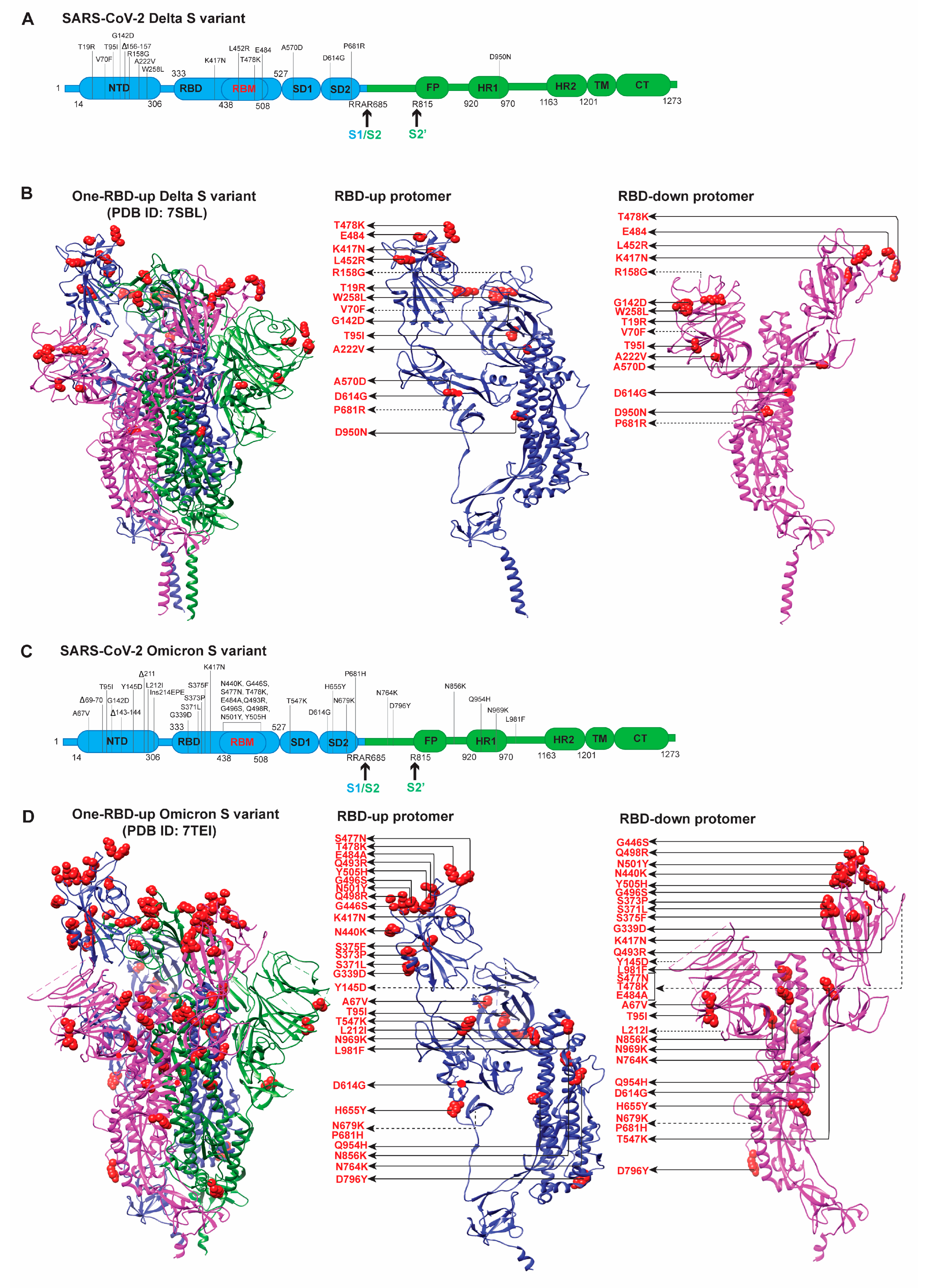
3.2. Delta/B.1.617.2 Variant
3.3. Omicron/B.1.1.529 Variant
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats Are Natural Reservoirs of SARS-like Coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Gorbalena, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The Species Severe Acute Respiratory Syndrome-Related Coronavirus: Classifying 2019-NCoV and Naming It SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Rabaan, A.A.; Al-Ahmed, S.H.; Haque, S.; Sah, R.; Tiwari, R.; Malik, Y.S.; Dhama, K.; Yatoo, M.I.; Bonilla-Aldana, D.K.; Rodriguez-Morales, A.J. SARS-CoV-2, SARS-CoV, and MERS-COV: A Comparative Overview. Infez. Med. 2020, 28, 174–184. [Google Scholar] [PubMed]
- Liu, D.X.; Liang, J.Q.; Fung, T.S. Human Coronavirus-229E, -OC43, -NL63, and -HKU1 (Coronaviridae). Encycl. Virol. 2021, 2, 428–440. [Google Scholar] [CrossRef]
- Pyrc, K.; Berkhout, B.; van der Hoek, L. The Novel Human Coronaviruses NL63 and HKU1. J. Virol. 2007, 81, 3051–3057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehr, A.R.; Perlman, S. Coronaviruses: An Overview of Their Replication and Pathogenesis. In Coronaviruses: Methods and Protocols; Maier, H.J., Bickerton, E., Britton, P., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2015; pp. 1–23. ISBN 978-1-4939-2438-7. [Google Scholar]
- Wang, Y.; Grunewald, M.; Perlman, S. Coronaviruses: An Updated Overview of Their Replication and Pathogenesis. Methods Mol. Biol. 2020, 2203, 1–29. [Google Scholar] [CrossRef]
- Bradburne, A.F.; Bynoe, M.L.; Tyrrell, D.A. Effects of a “New” Human Respiratory Virus in Volunteers. Br. Med. J. 1967, 3, 767–769. [Google Scholar] [CrossRef] [Green Version]
- Hamre, D.; Procknow, J.J. A New Virus Isolated from the Human Respiratory Tract. Proc. Soc. Exp. Biol. Med. 1966, 121, 190–193. [Google Scholar] [CrossRef]
- van der Hoek, L.; Pyrc, K.; Jebbink, M.F.; Vermeulen-Oost, W.; Berkhout, R.J.M.; Wolthers, K.C.; Wertheim-van Dillen, P.M.E.; Kaandorp, J.; Spaargaren, J.; Berkhout, B. Identification of a New Human Coronavirus. Nat. Med. 2004, 10, 368–373. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Lau, S.K.P.; Chu, C.; Chan, K.; Tsoi, H.; Huang, Y.; Wong, B.H.L.; Poon, R.W.S.; Cai, J.J.; Luk, W.; et al. Characterization and Complete Genome Sequence of a Novel Coronavirus, Coronavirus HKU1, from Patients with Pneumonia. J. Virol. 2005, 79, 884–895. [Google Scholar] [CrossRef] [Green Version]
- Vabret, A.; Mourez, T.; Gouarin, S.; Petitjean, J.; Freymuth, F. An Outbreak of Coronavirus OC43 Respiratory Infection in Normandy, France. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2003, 36, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Zhong, N.; Zheng, B.; Li, Y.; Poon, L.; Xie, Z.; Chan, K.; Li, P.; Tan, S.; Chang, Q.; Xie, J.; et al. Epidemiology and Cause of Severe Acute Respiratory Syndrome (SARS) in Guangdong, People’s Republic of China, in February, 2003. Lancet 2003, 362, 1353–1358. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic Characterisation and Epidemiology of 2019 Novel Coronavirus: Implications for Virus Origins and Receptor Binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.-Y.; Zhao, R.; Gao, L.-J.; Gao, X.-F.; Wang, D.-P.; Cao, J.-M. SARS-CoV-2: Structure, Biology, and Structure-Based Therapeutics Development. Front. Cell. Infect. Microbiol. 2020, 10, 587269. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome Composition and Divergence of the Novel Coronavirus (2019-NCoV) Originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef] [Green Version]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, M.I. Insights into SARS-CoV-2 Genome, Structure, Evolution, Pathogenesis and Therapies: Structural Genomics Approach. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef]
- Banerjee, A.K.; Blanco, M.R.; Bruce, E.A.; Honson, D.D.; Chen, L.M.; Chow, A.; Bhat, P.; Ollikainen, N.; Quinodoz, S.A.; Loney, C.; et al. SARS-CoV-2 Disrupts Splicing, Translation, and Protein Trafficking to Suppress Host Defenses. Cell 2020, 183, 1325–1339.e21. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell Entry Mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784.e5. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-Specific Glycan Analysis of the SARS-CoV-2 Spike. Science 2020, 369, 330–333. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.-Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM Structure of the 2019-NCoV Spike in the Prefusion Conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Huang, I.-C.; Bosch, B.J.; Li, F.; Li, W.; Lee, K.H.; Ghiran, S.; Vasilieva, N.; Dermody, T.S.; Harrison, S.C.; Dormitzer, P.R.; et al. SARS Coronavirus, but Not Human Coronavirus NL63, Utilizes Cathepsin L to Infect ACE2-Expressing Cells. J. Biol. Chem. 2006, 281, 3198–3203. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.-M.; Yang, W.-L.; Yang, F.-Y.; Zhang, L.; Huang, W.-J.; Hou, W.; Fan, C.-F.; Jin, R.-H.; Feng, Y.-M.; Wang, Y.-C.; et al. Cathepsin L Plays a Key Role in SARS-CoV-2 Infection in Humans and Humanized Mice and Is a Promising Target for New Drug Development. Signal Transduct. Target. Ther. 2021, 6, 134. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.-F.; Xu, W.; Liu, S.-W. Structural and Functional Properties of SARS-CoV-2 Spike Protein: Potential Antivirus Drug Development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Lv, Z.; Deng, Y.-Q.; Ye, Q.; Cao, L.; Sun, C.-Y.; Fan, C.; Huang, W.; Sun, S.; Sun, Y.; Zhu, L.; et al. Structural Basis for Neutralization of SARS-CoV-2 and SARS-CoV by a Potent Therapeutic Antibody. Science 2020, 369, 1505–1509. [Google Scholar] [CrossRef]
- Aoe, T. Pathological Aspects of COVID-19 as a Conformational Disease and the Use of Pharmacological Chaperones as a Potential Therapeutic Strategy. Front. Pharmacol. 2020, 11, 1095. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the MRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 MRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Voysey, M.; Clemens, S.A.C.; Madhi, S.A.; Weckx, L.Y.; Folegatti, P.M.; Aley, P.K.; Angus, B.; Baillie, V.L.; Barnabas, S.L.; Bhorat, Q.E.; et al. Safety and Efficacy of the ChAdOx1 NCoV-19 Vaccine (AZD1222) against SARS-CoV-2: An Interim Analysis of Four Randomised Controlled Trials in Brazil, South Africa, and the UK. Lancet 2021, 397, 99–111. [Google Scholar] [CrossRef]
- Sadoff, J.; Le Gars, M.; Shukarev, G.; Heerwegh, D.; Truyers, C.; de Groot, A.M.; Stoop, J.; Tete, S.; Van Damme, W.; Leroux-Roels, I.; et al. Interim Results of a Phase 1–2a Trial of Ad26.COV2.S Covid-19 Vaccine. N. Engl. J. Med. 2021, 384, 1824–1835. [Google Scholar] [CrossRef]
- Lloyd, E.C.; Gandhi, T.N.; Petty, L.A. Monoclonal Antibodies for COVID-19. JAMA 2021, 325, 1015. [Google Scholar] [CrossRef]
- Marovich, M.; Mascola, J.R.; Cohen, M.S. Monoclonal Antibodies for Prevention and Treatment of COVID-19. JAMA 2020, 324, 131–132. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 Spike Receptor-Binding Domain Bound to the ACE2 Receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural Basis of Receptor Recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural Basis for the Recognition of SARS-CoV-2 by Full-Length Human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Tsybovsky, Y.; Gorman, J.; Rapp, M.; Cerutti, G.; Chuang, G.-Y.; Katsamba, P.S.; Sampson, J.M.; Schön, A.; Bimela, J.; et al. Cryo-EM Structures of SARS-CoV-2 Spike without and with ACE2 Reveal a PH-Dependent Switch to Mediate Endosomal Positioning of Receptor-Binding Domains. Cell Host Microbe 2020, 28, 867–879.e5. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct Conformational States of SARS-CoV-2 Spike Protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef] [PubMed]
- Bangaru, S.; Ozorowski, G.; Turner, H.L.; Antanasijevic, A.; Huang, D.; Wang, X.; Torres, J.L.; Diedrich, J.K.; Tian, J.-H.; Portnoff, A.D.; et al. Structural Analysis of Full-Length SARS-CoV-2 Spike Protein from an Advanced Vaccine Candidate. Science 2020, 370, 1089–1094. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Song, Y.; Chen, Y.; Wu, N.; Xu, J.; Sun, C.; Zhang, J.; Weng, T.; Zhang, Z.; Wu, Z.; et al. Molecular Architecture of the SARS-CoV-2 Virus. Cell 2020, 183, 730–738.e13. [Google Scholar] [CrossRef]
- Ke, Z.; Oton, J.; Qu, K.; Cortese, M.; Zila, V.; McKeane, L.; Nakane, T.; Zivanov, J.; Neufeldt, C.J.; Cerikan, B.; et al. Structures and Distributions of SARS-CoV-2 Spike Proteins on Intact Virions. Nature 2020, 588, 498–502. [Google Scholar] [CrossRef]
- Turoňová, B.; Sikora, M.; Schürmann, C.; Hagen, W.J.H.; Welsch, S.; Blanc, F.E.C.; von Bülow, S.; Gecht, M.; Bagola, K.; Hörner, C.; et al. In Situ Structural Analysis of SARS-CoV-2 Spike Reveals Flexibility Mediated by Three Hinges. Science 2020, 370, 203–208. [Google Scholar] [CrossRef]
- Liu, C.; Mendonça, L.; Yang, Y.; Gao, Y.; Shen, C.; Liu, J.; Ni, T.; Ju, B.; Liu, C.; Tang, X.; et al. The Architecture of Inactivated SARS-CoV-2 with Postfusion Spikes Revealed by Cryo-EM and Cryo-ET. Structure 2020, 28, 1218. [Google Scholar] [CrossRef]
- Cueno, M.E.; Imai, K. Structural Comparison of the SARS CoV 2 Spike Protein Relative to Other Human-Infecting Coronaviruses. Front. Med. 2021, 7, 594439. [Google Scholar] [CrossRef]
- Kumar, V.G.; Ogden, D.S.; Isu, U.H.; Polasa, A.; Losey, J.; Moradi, M. Prefusion Spike Protein Conformational Changes Are Slower in SARS-CoV-2 than in SARS-CoV-1. J. Biol. Chem. 2022, 298, 101814. [Google Scholar] [CrossRef]
- Wrobel, A.G.; Benton, D.J.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. SARS-CoV-2 and Bat RaTG13 Spike Glycoprotein Structures Inform on Virus Evolution and Furin-Cleavage Effects. Nat. Struct. Mol. Biol. 2020, 27, 763–767. [Google Scholar] [CrossRef]
- Cao, L.; Goreshnik, I.; Coventry, B.; Case, J.B.; Miller, L.; Kozodoy, L.; Chen, R.E.; Carter, L.; Walls, A.C.; Park, Y.-J.; et al. De Novo Design of Picomolar SARS-CoV-2 Miniprotein Inhibitors. Science 2020, 370, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Uchil, P.D.; Li, W.; Zheng, D.; Terry, D.S.; Gorman, J.; Shi, W.; Zhang, B.; Zhou, T.; Ding, S.; et al. Real-Time Conformational Dynamics of SARS-CoV-2 Spikes on Virus Particles. Cell Host Microbe 2020, 28, 880–891.e8. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Han, Y.; Ding, S.; Shi, W.; Zhou, T.; Finzi, A.; Kwong, P.D.; Mothes, W.; Lu, M. SARS-CoV-2 Variants Increase Kinetic Stability of Open Spike Conformations as an Evolutionary Strategy. mBio 2022, 13, e0322721. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cai, Y.; Xiao, T.; Lu, J.; Peng, H.; Sterling, S.M.; Walsh, R.M.; Rits-Volloch, S.; Zhu, H.; Woosley, A.N.; et al. Structural Impact on SARS-CoV-2 Spike Protein by D614G Substitution. Science 2021, 372, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Benton, D.J.; Wrobel, A.G.; Roustan, C.; Borg, A.; Xu, P.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. The Effect of the D614G Substitution on the Structure of the Spike Glycoprotein of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2021, 118, e2022586118. [Google Scholar] [CrossRef] [PubMed]
- Gobeil, S.M.-C.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Manne, K.; Stalls, V.; Kopp, M.F.; Henderson, R.; Edwards, R.J.; et al. D614G Mutation Alters SARS-CoV-2 Spike Conformation and Enhances Protease Cleavage at the S1/S2 Junction. Cell Rep. 2021, 34, 108630. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; et al. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell 2020, 183, 739–751.e8. [Google Scholar] [CrossRef]
- Díaz-Salinas, M.A.; Li, Q.; Ejemel, M.; Yurkovetskiy, L.; Luban, J.; Shen, K.; Wang, Y.; Munro, J.B. Conformational Dynamics and Allosteric Modulation of the SARS-CoV-2 Spike. eLife 2022, 11, e75433. [Google Scholar] [CrossRef]
- CDC Coronavirus Disease 2019 (COVID-19). Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-classifications.html (accessed on 23 December 2021).
- Yi, H.; Wang, J.; Wang, J.; Lu, Y.; Zhang, Y.; Peng, R.; Lu, J.; Chen, Z. The Emergence and Spread of Novel SARS-CoV-2 Variants. Front. Public Health 2021, 9, 1017. [Google Scholar] [CrossRef]
- SARS-CoV-2 Variants of Concern as of 22 December 2021. Available online: https://www.ecdc.europa.eu/en/covid-19/variants-concern (accessed on 23 December 2021).
- Cameroni, E.; Bowen, J.E.; Rosen, L.E.; Saliba, C.; Zepeda, S.K.; Culap, K.; Pinto, D.; VanBlargan, L.A.; De Marco, A.; di Iulio, J.; et al. Broadly Neutralizing Antibodies Overcome SARS-CoV-2 Omicron Antigenic Shift. Nature 2021, 602, 664–670. [Google Scholar] [CrossRef]
- McCallum, M.; Bassi, J.; De Marco, A.; Chen, A.; Walls, A.C.; Di Iulio, J.; Tortorici, M.A.; Navarro, M.-J.; Silacci-Fregni, C.; Saliba, C.; et al. SARS-CoV-2 Immune Evasion by the B.1.427/B.1.429 Variant of Concern. Science 2021, 373, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wang, J.; Jian, F.; Xiao, T.; Song, W.; Yisimayi, A.; Huang, W.; Li, Q.; Wang, P.; An, R.; et al. Omicron Escapes the Majority of Existing SARS-CoV-2 Neutralizing Antibodies. Nature 2021, 602, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.; Logan, N.; Tyson, G.; Orton, R.; Harvey, W.T.; Perkins, J.S.; Mollett, G.; Blacow, R.M.; The COVID-19 Genomics UK (COG-UK) Consortium; Peacock, T.P.; et al. Reduced Neutralisation of the Delta (B.1.617.2) SARS-CoV-2 Variant of Concern Following Vaccination. PLoS Pathog. 2021, 17, e1010022. [Google Scholar] [CrossRef] [PubMed]
- Muik, A.; Wallisch, A.-K.; Sänger, B.; Swanson, K.A.; Mühl, J.; Chen, W.; Cai, H.; Maurus, D.; Sarkar, R.; Türeci, Ö.; et al. Neutralization of SARS-CoV-2 Lineage B.1.1.7 Pseudovirus by BNT162b2 Vaccine-Elicited Human Sera. Science 2021, 371, 1152–1153. [Google Scholar] [CrossRef] [PubMed]
- Fernández, J.; Bruneau, N.; Fasce, R.; Martín, H.S.; Balanda, M.; Bustos, P.; Ulloa, S.; Mora, J.; Ramírez, E. Neutralization of Alpha, Gamma, and D614G SARS-CoV-2 Variants by CoronaVac Vaccine-Induced Antibodies. J. Med. Virol. 2022, 94, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced Sensitivity of SARS-CoV-2 Variant Delta to Antibody Neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef]
- Supasa, P.; Zhou, D.; Dejnirattisai, W.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.E.; Nutalai, R.; Tuekprakhon, A.; et al. Reduced Neutralization of SARS-CoV-2 B.1.1.7 Variant by Convalescent and Vaccine Sera. Cell 2021, 184, 2201–2211.e7. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, J.; Xiao, T.; Lavine, C.L.; Rawson, S.; Peng, H.; Zhu, H.; Anand, K.; Tong, P.; Gautam, A.; et al. Structural Basis for Enhanced Infectivity and Immune Evasion of SARS-CoV-2 Variants. Science 2021, 373, 642–648. [Google Scholar] [CrossRef]
- Bowen, J.E.; Walls, A.C.; Joshi, A.; Sprouse, K.R.; Stewart, C.; Tortorici, M.A.; Franko, N.M.; Logue, J.K.; Mazzitelli, I.G.; Tiles, S.W.; et al. SARS-CoV-2 Spike Conformation Determines Plasma Neutralizing Activity. BioRxiv 2021. [Google Scholar] [CrossRef]
- Li, W.; Chen, Y.; Prévost, J.; Ullah, I.; Lu, M.; Gong, S.Y.; Tauzin, A.; Gasser, R.; Vézina, D.; Anand, S.P.; et al. Structural Basis and Mode of Action for Two Broadly Neutralizing Antibodies Against SARS-CoV-2 Emerging Variants of Concern. Cell Rep. 2021, 38, 110210. [Google Scholar] [CrossRef]
- Wang, P.; Nair, M.S.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; et al. Antibody Resistance of SARS-CoV-2 Variants B.1.351 and B.1.1.7. BioRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Iketani, S.; Liu, L.; Guo, Y.; Liu, L.; Chan, J.F.-W.; Huang, Y.; Wang, M.; Luo, Y.; Yu, J.; Chu, H.; et al. Antibody Evasion Properties of SARS-CoV-2 Omicron Sublineages. Nature 2022, 604, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xiao, T.; Cai, Y.; Lavine, C.L.; Peng, H.; Zhu, H.; Anand, K.; Tong, P.; Gautam, A.; Mayer, M.L.; et al. Membrane Fusion and Immune Evasion by the Spike Protein of SARS-CoV-2 Delta Variant. Science 2021, 374, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Papa, G.; Mallery, D.L.; Albecka, A.; Welch, L.G.; Cattin-Ortolá, J.; Luptak, J.; Paul, D.; McMahon, H.T.; Goodfellow, I.G.; Carter, A.; et al. Furin Cleavage of SARS-CoV-2 Spike Promotes but Is Not Essential for Infection and Cell-Cell Fusion. PLoS Pathog. 2021, 17, e1009246. [Google Scholar] [CrossRef]
- Shen, X.; Tang, H.; McDanal, C.; Wagh, K.; Fischer, W.; Theiler, J.; Yoon, H.; Li, D.; Haynes, B.F.; Sanders, K.O.; et al. SARS-CoV-2 Variant B.1.1.7 Is Susceptible to Neutralizing Antibodies Elicited by Ancestral Spike Vaccines. Cell Host Microbe 2021, 29, 529–539.e3. [Google Scholar] [CrossRef]
- Collier, D.A.; De Marco, A.; Ferreira, I.A.T.M.; Meng, B.; Datir, R.P.; Walls, A.C.; Kemp, S.A.; Bassi, J.; Pinto, D.; Silacci-Fregni, C.; et al. Sensitivity of SARS-CoV-2 B.1.1.7 to MRNA Vaccine-Elicited Antibodies. Nature 2021, 593, 136–141. [Google Scholar] [CrossRef]
- Graham, C.; Seow, J.; Huettner, I.; Khan, H.; Kouphou, N.; Acors, S.; Winstone, H.; Pickering, S.; Galao, R.P.; Dupont, L.; et al. Neutralization Potency of Monoclonal Antibodies Recognizing Dominant and Subdominant Epitopes on SARS-CoV-2 Spike Is Impacted by the B.1.1.7 Variant. Immunity 2021, 54, 1276–1289.e6. [Google Scholar] [CrossRef]
- Zhou, D.; Dejnirattisai, W.; Supasa, P.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.E.; Tuekprakhon, A.; Nutalai, R.; et al. Evidence of Escape of SARS-CoV-2 Variant B.1.351 from Natural and Vaccine-Induced Sera. Cell 2021, 184, 2348–2361.e6. [Google Scholar] [CrossRef]
- Souza, W.M.; Amorim, M.R.; Sesti-Costa, R.; Coimbra, L.D.; Brunetti, N.S.; Toledo-Teixeira, D.A.; de Souza, G.F.; Muraro, S.P.; Parise, P.L.; Barbosa, P.P.; et al. Neutralisation of SARS-CoV-2 Lineage P.1 by Antibodies Elicited through Natural SARS-CoV-2 Infection or Vaccination with an Inactivated SARS-CoV-2 Vaccine: An Immunological Study. Lancet Microbe 2021, 2, e527–e535. [Google Scholar] [CrossRef]
- Mlcochova, P.; Kemp, S.A.; Dhar, M.S.; Papa, G.; Meng, B.; Ferreira, I.A.T.M.; Datir, R.; Collier, D.A.; Albecka, A.; Singh, S.; et al. SARS-CoV-2 B.1.617.2 Delta Variant Replication and Immune Evasion. Nature 2021, 599, 114–119. [Google Scholar] [CrossRef]
- Cele, S.; Jackson, L.; Khoury, D.S.; Khan, K.; Moyo-Gwete, T.; Tegally, H.; San, J.E.; Cromer, D.; Scheepers, C.; Amoako, D.; et al. Omicron Extensively but Incompletely Escapes Pfizer BNT162b2 Neutralization. Nature 2021, 602, 654–656. [Google Scholar] [CrossRef] [PubMed]
- Planas, D.; Saunders, N.; Maes, P.; Guivel-Benhassine, F.; Planchais, C.; Buchrieser, J.; Bolland, W.-H.; Porrot, F.; Staropoli, I.; Lemoine, F.; et al. Considerable Escape of SARS-CoV-2 Omicron to Antibody Neutralization. Nature 2021, 602, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Nemet, I.; Kliker, L.; Lustig, Y.; Zuckerman, N.; Erster, O.; Cohen, C.; Kreiss, Y.; Alroy-Preis, S.; Regev-Yochay, G.; Mendelson, E.; et al. Third BNT162b2 Vaccination Neutralization of SARS-CoV-2 Omicron Infection. N. Engl. J. Med. 2022, 386, 492–494. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Iketani, S.; Guo, Y.; Chan, J.F.-W.; Wang, M.; Liu, L.; Luo, Y.; Chu, H.; Huang, Y.; Nair, M.S.; et al. Striking Antibody Evasion Manifested by the Omicron Variant of SARS-CoV-2. Nature 2021, 602, 676–681. [Google Scholar] [CrossRef]
- Hastie, K.M.; Li, H.; Bedinger, D.; Schendel, S.L.; Dennison, S.M.; Li, K.; Rayaprolu, V.; Yu, X.; Mann, C.; Zandonatti, M.; et al. Defining Variant-Resistant Epitopes Targeted by SARS-CoV-2 Antibodies: A Global Consortium Study. Science 2021, 374, 472–478. [Google Scholar] [CrossRef]
- Gobeil, S.M.-C.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Stalls, V.; Kopp, M.F.; Manne, K.; Li, D.; Wiehe, K.; et al. Effect of Natural Mutations of SARS-CoV-2 on Spike Structure, Conformation, and Antigenicity. Science 2021, 373, eabi6226. [Google Scholar] [CrossRef]
- Bocková, M.; Slabý, J.; Špringer, T.; Homola, J. Advances in Surface Plasmon Resonance Imaging and Microscopy and Their Biological Applications. Annu. Rev. Anal. Chem. 2019, 12, 151–176. [Google Scholar] [CrossRef]
- Alhajj, M.; Farhana, A. Enzyme Linked Immunosorbent Assay; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Sultana, A.; Lee, J.E. Measuring Protein-Protein and Protein-Nucleic Acid Interactions by Biolayer Interferometry. Curr. Protoc. Protein Sci. 2015, 79, 19.25.1–19.25.26. [Google Scholar] [CrossRef]
- Tauzin, A.; Nayrac, M.; Benlarbi, M.; Gong, S.Y.; Gasser, R.; Beaudoin-Bussières, G.; Brassard, N.; Laumaea, A.; Vézina, D.; Prévost, J.; et al. A Single Dose of the SARS-CoV-2 Vaccine BNT162b2 Elicits Fc-Mediated Antibody Effector Functions and T Cell Responses. Cell Host Microbe 2021, 29, 1137–1150.e6. [Google Scholar] [CrossRef]
- Prévost, J.; Gasser, R.; Beaudoin-Bussières, G.; Richard, J.; Duerr, R.; Laumaea, A.; Anand, S.P.; Goyette, G.; Benlarbi, M.; Ding, S.; et al. Cross-Sectional Evaluation of Humoral Responses against SARS-CoV-2 Spike. Cell Rep. Med. 2020, 1, 100126. [Google Scholar] [CrossRef]
- Jaafar, R.; Boschi, C.; Aherfi, S.; Bancod, A.; Le Bideau, M.; Edouard, S.; Colson, P.; Chahinian, H.; Raoult, D.; Yahi, N.; et al. High Individual Heterogeneity of Neutralizing Activities against the Original Strain and Nine Different Variants of SARS-CoV-2. Viruses 2021, 13, 2177. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Arora, P.; Groß, R.; Seidel, A.; Hörnich, B.F.; Hahn, A.S.; Krüger, N.; Graichen, L.; Hofmann-Winkler, H.; Kempf, A.; et al. SARS-CoV-2 Variants B.1.351 and P.1 Escape from Neutralizing Antibodies. Cell 2021, 184, 2384–2393.e12. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Casner, R.G.; Nair, M.S.; Wang, M.; Yu, J.; Cerutti, G.; Liu, L.; Kwong, P.D.; Huang, Y.; Shapiro, L.; et al. Increased Resistance of SARS-CoV-2 Variant P.1 to Antibody Neutralization. Cell Host Microbe 2021, 29, 747–751.e4. [Google Scholar] [CrossRef]
- Liu, C.; Ginn, H.M.; Dejnirattisai, W.; Supasa, P.; Wang, B.; Tuekprakhon, A.; Nutalai, R.; Zhou, D.; Mentzer, A.J.; Zhao, Y.; et al. Reduced Neutralization of SARS-CoV-2 B.1.617 by Vaccine and Convalescent Serum. Cell 2021, 184, 4220–4236.e13. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, C.; Zhang, C.; Wang, Y.; Hong, Q.; Xu, S.; Li, Z.; Yang, Y.; Huang, Z.; Cong, Y. Structural Basis for SARS-CoV-2 Delta Variant Recognition of ACE2 Receptor and Broadly Neutralizing Antibodies. Nat. Commun. 2022, 13, 871. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.; Muecksch, F.; Weisblum, Y.; Da Silva, J.; Bednarski, E.; Cho, A.; Wang, Z.; Gaebler, C.; Caskey, M.; Nussenzweig, M.C.; et al. Plasma Neutralization of the SARS-CoV-2 Omicron Variant. N. Engl. J. Med. 2022, 386, 599–601. [Google Scholar] [CrossRef]
- McCallum, M.; Czudnochowski, N.; Rosen, L.E.; Zepeda, S.K.; Bowen, J.E.; Walls, A.C.; Hauser, K.; Joshi, A.; Stewart, C.; Dillen, J.R.; et al. Structural Basis of SARS-CoV-2 Omicron Immune Evasion and Receptor Engagement. Science 2022, 375, 864–868. [Google Scholar] [CrossRef]
- Mannar, D.; Saville, J.W.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Tuttle, K.S.; Marquez, A.C.; Sekirov, I.; Subramaniam, S. SARS-CoV-2 Omicron Variant: Antibody Evasion and Cryo-EM Structure of Spike Protein–ACE2 Complex. Science 2022, 375, 760–764. [Google Scholar] [CrossRef]
- Yin, W.; Xu, Y.; Xu, P.; Cao, X.; Wu, C.; Gu, C.; He, X.; Wang, X.; Huang, S.; Yuan, Q.; et al. Structures of the Omicron Spike Trimer with ACE2 and an Anti-Omicron Antibody. Science 2022, 375, 1048–1053. [Google Scholar] [CrossRef]
- Zhang, J.; Cai, Y.; Lavine, C.L.; Peng, H.; Zhu, H.; Anand, K.; Tong, P.; Gautam, A.; Mayer, M.L.; Rits-Volloch, S.; et al. Structural and Functional Impact by SARS-CoV-2 Omicron Spike Mutations. Cell Rep. 2022, 39, 110729. [Google Scholar] [CrossRef]
- Lazarevic, I.; Pravica, V.; Miljanovic, D.; Cupic, M. Immune Evasion of SARS-CoV-2 Emerging Variants: What Have We Learnt So Far? Viruses 2021, 13, 1192. [Google Scholar] [CrossRef]
- Rees-Spear, C.; Muir, L.; Griffith, S.A.; Heaney, J.; Aldon, Y.; Snitselaar, J.L.; Thomas, P.; Graham, C.; Seow, J.; Lee, N.; et al. The Effect of Spike Mutations on SARS-CoV-2 Neutralization. Cell Rep. 2021, 34, 108890. [Google Scholar] [CrossRef]
- Yang, T.-J.; Yu, P.-Y.; Chang, Y.-C.; Liang, K.-H.; Tso, H.-C.; Ho, M.-R.; Chen, W.-Y.; Lin, H.-T.; Wu, H.-C.; Hsu, S.-T.D. Effect of SARS-CoV-2 B.1.1.7 Mutations on Spike Protein Structure and Function. Nat. Struct. Mol. Biol. 2021, 28, 731–739. [Google Scholar] [CrossRef]
- Zahradník, J.; Marciano, S.; Shemesh, M.; Zoler, E.; Harari, D.; Chiaravalli, J.; Meyer, B.; Rudich, Y.; Li, C.; Marton, I.; et al. SARS-CoV-2 Variant Prediction and Antiviral Drug Design Are Enabled by RBD in Vitro Evolution. Nat. Microbiol. 2021, 6, 1188–1198. [Google Scholar] [CrossRef]
- Yang, T.-J.; Yu, P.-Y.; Chang, Y.-C.; Chang, N.-E.; Tsai, Y.-X.; Liang, K.-H.; Draczkowski, P.; Lin, B.; Wang, Y.-S.; Chien, Y.-C.; et al. Structure-Activity Relationships of B.1.617 and Other SARS-CoV-2 Spike Variants. BioRxiv 2021. [Google Scholar] [CrossRef]
- Shen, X.; Tang, H.; Pajon, R.; Smith, G.; Glenn, G.M.; Shi, W.; Korber, B.; Montefiori, D.C. Neutralization of SARS-CoV-2 Variants B.1.429 and B.1.351. N. Engl. J. Med. 2021, 384, 2352–2354. [Google Scholar] [CrossRef]
- Corti, D.; Purcell, L.A.; Snell, G.; Veesler, D. Tackling COVID-19 with Neutralizing Monoclonal Antibodies. Cell 2021, 184, 3086–3108. [Google Scholar] [CrossRef]
- Pinto, D.; Park, Y.-J.; Beltramello, M.; Walls, A.C.; Tortorici, M.A.; Bianchi, S.; Jaconi, S.; Culap, K.; Zatta, F.; De Marco, A.; et al. Cross-Neutralization of SARS-CoV-2 by a Human Monoclonal SARS-CoV Antibody. Nature 2020, 583, 290–295. [Google Scholar] [CrossRef]
- Taylor, C.A. COVID-19–Associated Hospitalizations Among Adults During SARS-CoV-2 Delta and Omicron Variant Predominance, by Race/Ethnicity and Vaccination Status—COVID-NET, 14 States, July 2021–January 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 466–473. [Google Scholar] [CrossRef]
- Nyberg, T.; Ferguson, N.M.; Nash, S.G.; Webster, H.H.; Flaxman, S.; Andrews, N.; Hinsley, W.; Bernal, J.L.; Kall, M.; Bhatt, S.; et al. Comparative Analysis of the Risks of Hospitalisation and Death Associated with SARS-CoV-2 Omicron (B.1.1.529) and Delta (B.1.617.2) Variants in England: A Cohort Study. Lancet 2022, 399, 1303–1312. [Google Scholar] [CrossRef]
- Shuai, H.; Chan, J.F.-W.; Hu, B.; Chai, Y.; Yuen, T.T.-T.; Yin, F.; Huang, X.; Yoon, C.; Hu, J.-C.; Liu, H.; et al. Attenuated Replication and Pathogenicity of SARS-CoV-2 B.1.1.529 Omicron. Nature 2022, 603, 693–699. [Google Scholar] [CrossRef]
- Lamers, M.M.; Mykytyn, A.Z.; Breugem, T.I.; Groen, N.; Knoops, K.; Schipper, D.; van Acker, R.; van den Doel, P.B.; Bestebroer, T.; Koopman, C.D.; et al. SARS-CoV-2 Omicron Efficiently Infects Human Airway, but Not Alveolar Epithelium. BioRxiv 2022. [Google Scholar] [CrossRef]
- Halfmann, P.J.; Iida, S.; Iwatsuki-Horimoto, K.; Maemura, T.; Kiso, M.; Scheaffer, S.M.; Darling, T.L.; Joshi, A.; Loeber, S.; Singh, G.; et al. SARS-CoV-2 Omicron Virus Causes Attenuated Disease in Mice and Hamsters. Nature 2022, 603, 687–692. [Google Scholar] [CrossRef]
- Fantini, J.; Yahi, N.; Azzaz, F.; Chahinian, H. Structural Dynamics of SARS-CoV-2 Variants: A Health Monitoring Strategy for Anticipating Covid-19 Outbreaks. J. Infect. 2021, 83, 197–206. [Google Scholar] [CrossRef]
- Du, S.; Liu, P.; Zhang, Z.; Xiao, T.; Yasimayi, A.; Huang, W.; Wang, Y.; Cao, Y.; Xie, X.S.; Xiao, J. Structures of SARS-CoV-2 B.1.351 Neutralizing Antibodies Provide Insights into Cocktail Design against Concerning Variants. Cell Res. 2021, 31, 1130–1133. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, C.; Wang, Y.; Hong, Q.; Zhang, C.; Li, Z.; Xu, S.; Zuo, Q.; Liu, C.; Huang, Z.; et al. Conformational Dynamics of the Beta and Kappa SARS-CoV-2 Spike Proteins and Their Complexes with ACE2 Receptor Revealed by Cryo-EM. Nat. Commun. 2021, 12, 7345. [Google Scholar] [CrossRef]
- Villoutreix, B.O.; Calvez, V.; Marcelin, A.-G.; Khatib, A.-M. In Silico Investigation of the New UK (B.1.1.7) and South African (501Y.V2) SARS-CoV-2 Variants with a Focus at the ACE2–Spike RBD Interface. Int. J. Mol. Sci. 2021, 22, 1695. [Google Scholar] [CrossRef]
- Ortega, J.T.; Serrano, M.L.; Pujol, F.H.; Rangel, H.R. Role of Changes in SARS-CoV-2 Spike Protein in the Interaction with the Human ACE2 Receptor: An in Silico Analysis. EXCLI J. 2020, 19, 410–417. [Google Scholar] [CrossRef]
- Ortega, J.T.; Jastrzebska, B.; Rangel, H.R. Omicron SARS-CoV-2 Variant Spike Protein Shows an Increased Affinity to the Human ACE2 Receptor: An In Silico Analysis. Pathogens 2022, 11, 45. [Google Scholar] [CrossRef]
- Verma, J.; Subbarao, N. Insilico Study on the Effect of SARS-CoV-2 RBD Hotspot Mutants’ Interaction with ACE2 to Understand the Binding Affinity and Stability. Virology 2021, 561, 107–116. [Google Scholar] [CrossRef]
- McCallum, M.; Walls, A.C.; Sprouse, K.R.; Bowen, J.E.; Rosen, L.E.; Dang, H.V.; De Marco, A.; Franko, N.; Tilles, S.W.; Logue, J.; et al. Molecular Basis of Immune Evasion by the Delta and Kappa SARS-CoV-2 Variants. Science 2021, 374, 1621–1626. [Google Scholar] [CrossRef]
- Han, P.; Li, L.; Liu, S.; Wang, Q.; Zhang, D.; Xu, Z.; Han, P.; Li, X.; Peng, Q.; Su, C.; et al. Receptor Binding and Complex Structures of Human ACE2 to Spike RBD from Omicron and Delta SARS-CoV-2. Cell 2022, 185, 630–640.e10. [Google Scholar] [CrossRef]
- Gobeil, S.M.-C.; Henderson, R.; Stalls, V.; Janowska, K.; Huang, X.; May, A.; Speakman, M.; Beaudoin, E.; Manne, K.; Li, D.; et al. Structural Diversity of the SARS-CoV-2 Omicron Spike. Mol. Cell 2022, 82, 2050–2068.e6. [Google Scholar] [CrossRef]
- Mannar, D.; Saville, J.W.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Zhou, S.; Tuttle, K.S.; Kim, A.; Li, W.; Dimitrov, D.S.; et al. Structural Analysis of Receptor Binding Domain Mutations in SARS-CoV-2 Variants of Concern That Modulate ACE2 and Antibody Binding. Cell Rep. 2021, 37, 110156. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SARS-CoV-2 Variants | Neutralizing Capacity | Binding Capacity of RBD with hACE2 | |||
|---|---|---|---|---|---|
| Plasma from Convalescent Patient | Sera from Vaccinated Individuals | RBD-Directed Antibodies | NTD-Directed Antibodies | ||
| Alpha | Slightly reduced: >1-fold (compared to D614G) [70,74,78,95] | Reduced: >2-fold (D614G) [67,70,74,78,95] | Reduced: wide range >1- to >100-fold to total loss [70,74,79,95] | Completely reduced: total loss of neutralization [74,79,80] | Comparable and noticeably increased: >2-fold (D614G) [70,89] |
| Beta | Reduced: >7-fold (D614G) [81,95,96] | Reduced: >7-fold (D614G) [81,95,96] | Reduced: wide range >1- to >100-fold to total loss (D614G) [81,95,96] | Completely reduced: Total loss of neutralization [96] | Increased: >15-fold (Victoria Strain) [81] |
| Gamma | Reduced: >3-fold (B lineage isolate) [82,95,96,97]. | Reduced: >3-fold (D614G) [82,96,97]. | Reduced: wide range >1- to >100-fold reduction to total loss (D614G) [95,96,97]. | Dramatically or completely reduced [82,96,97] | Increased: >18-fold (Victoria strain) [76,98] |
| Delta | Reduced: >5-fold (D614G) [83,95]. | Reduced: >5 to >10-fold (D614G WT), 3- to 5-fold (Alpha) [66,69,83]. | Reduced: wide range 2- to >40-fold to total abrogation (D614G) [69,83,95]. | Dramatically or completely reduced [69,83] | Increased: >10-fold (D614G) [76,99] |
| Omicron | Dramatically reduced: >70-fold to total loss (D614G) [63,75,84,85,86,87,100]. | Dramatically reduced: >35- to <60-fold (D614G) [63,75,84,85,86,100]. | Reduced: wide range >1- to >100-fold to total loss (D614G) [63,85,87,101] | Completely reduced: [63,101] | Increased: <10-fold (D614G) [63,102,103,104]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghimire, D.; Han, Y.; Lu, M. Structural Plasticity and Immune Evasion of SARS-CoV-2 Spike Variants. Viruses 2022, 14, 1255. https://doi.org/10.3390/v14061255
Ghimire D, Han Y, Lu M. Structural Plasticity and Immune Evasion of SARS-CoV-2 Spike Variants. Viruses. 2022; 14(6):1255. https://doi.org/10.3390/v14061255
Chicago/Turabian StyleGhimire, Dibya, Yang Han, and Maolin Lu. 2022. "Structural Plasticity and Immune Evasion of SARS-CoV-2 Spike Variants" Viruses 14, no. 6: 1255. https://doi.org/10.3390/v14061255
APA StyleGhimire, D., Han, Y., & Lu, M. (2022). Structural Plasticity and Immune Evasion of SARS-CoV-2 Spike Variants. Viruses, 14(6), 1255. https://doi.org/10.3390/v14061255