
Shiftless, a Critical Piece of the Innate Immune Response to Viral Infection


Abstract
1. Introduction
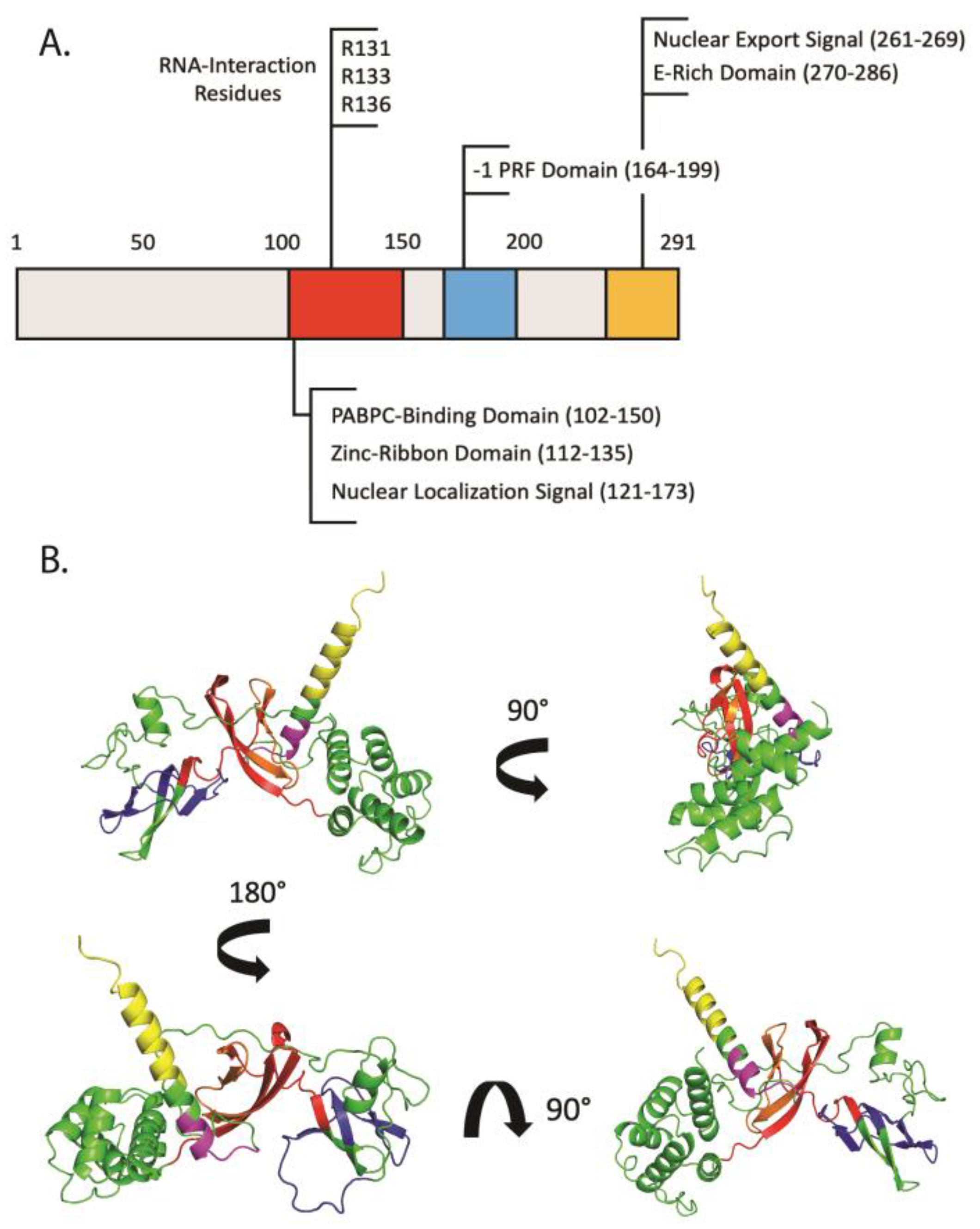
2. Shiftless: An ISG of Many Names
3. Shiftless and Viral RNA Fate
3.1. SHFL and Viral RNA Translation
3.2. Shiftless and Viral RNA Stabillity
3.3. Shiftless and RNA Granules during RNA Virus Infection
3.4. Shiftless and RNA Granules during DNA Virus Infection
4. Shiftless and the -1 Programmed Ribosomal Frameshift
5. Conclusions
Funding
Conflicts of Interest
References
- Schoggins, J.W. Interferon-Stimulated Genes: What Do They All Do? Annu. Rev. Virol. 2019, 6, 567–584. [Google Scholar] [CrossRef]
- Bull, T.M.; Meadows, C.A.; Coldren, C.D.; Moore, M.; Sotto-Santiago, S.M.; Nana-Sinkam, S.P.; Campbell, T.B.; Geraci, M.W. Human herpesvirus-8 infection of primary pulmonary microvascular endothelial cells. Am. J. Respir. Cell Mol. Biol. 2008, 39, 706–716. [Google Scholar] [CrossRef]
- Gaucher, D.; Therrien, R.; Kettaf, N.; Angermann, B.R.; Boucher, G.; Filali-Mouhim, A.; Moser, J.M.; Mehta, R.S.; Drake, D.R., III; Castro, E.; et al. Yellow fever vaccine induces integrated multilineage and polyfunctional immune responses. J. Exp. Med. 2008, 205, 3119–3131. [Google Scholar] [CrossRef]
- Harvey, S.A.; Romanowski, E.G.; Yates, K.A.; Gordon, Y.J. Adenovirus-directed ocular innate immunity: The role of conjunctival defensin-like chemokines (IP-10, I-TAC) and phagocytic human defensin-α. Investig. Ophthalmol. Vis. Sci. 2005, 46, 3657–3665. [Google Scholar] [CrossRef]
- Kash, J.C.; Mühlberger, E.; Carter, V.; Grosch, M.; Perwitasari, O.; Proll, S.C.; Thomas, M.J.; Weber, F.; Klenk, H.-D.; Katze, M.G. Global suppression of the host antiviral response by Ebola-and Marburgviruses: Increased antagonism of the type I interferon response is associated with enhanced virulence. J. Virol. 2006, 80, 3009–3020. [Google Scholar] [CrossRef]
- Miyazaki, D.; Haruki, T.; Takeda, S.; Sasaki, S.-I.; Yakura, K.; Terasaka, Y.; Komatsu, N.; Yamagami, S.; Touge, H.; Touge, C. Herpes simplex virus type 1–induced transcriptional networks of corneal endothelial cells indicate antigen presentation function. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4282–4293. [Google Scholar] [CrossRef][Green Version]
- Wang, J.; Nikrad, M.P.; Phang, T.; Gao, B.; Alford, T.; Ito, Y.; Edeen, K.; Travanty, E.A.; Kosmider, B.; Hartshorn, K. Innate immune response to influenza A virus in differentiated human alveolar type II cells. Am. J. Respir. Cell Mol. Biol. 2011, 45, 582–591. [Google Scholar] [CrossRef]
- Xiong, W.; Contreras, D.; Irudayam, J.I.; Ali, A.; Yang, O.O.; Arumugaswami, V. C19ORF66 is an interferon-stimulated gene (ISG) which Inhibits human immunodeficiency virus-1. bioRxiv 2016. [Google Scholar] [CrossRef]
- Zapata, J.C.; Carrion, R., Jr.; Patterson, J.L.; Crasta, O.; Zhang, Y.; Mani, S.; Jett, M.; Poonia, B.; Djavani, M.; White, D.M. Transcriptome analysis of human peripheral blood mononuclear cells exposed to Lassa virus and to the attenuated Mopeia/Lassa reassortant 29 (ML29), a vaccine candidate. PLoS Negl. Trop. Dis. 2013, 7, e2406. [Google Scholar] [CrossRef]
- Suzuki, Y.; Chin, W.X.; Han, Q.E.; Ichiyama, K.; Lee, C.H.; Eyo, Z.W.; Ebina, H.; Takahashi, H.; Takahashi, C.; Tan, B.H.; et al. Characterization of RyDEN (C19orf66) as an Interferon-Stimulated Cellular Inhibitor against Dengue Virus Replication. PLoS Pathog. 2016, 12, e1005357. [Google Scholar] [CrossRef]
- Balinsky, C.A.; Schmeisser, H.; Wells, A.I.; Ganesan, S.; Jin, T.; Singh, K.; Zoon, K.C. IRAV (FLJ11286), an Interferon-Stimulated Gene with Antiviral Activity against Dengue Virus, Interacts with MOV10. J. Virol. 2017, 91, e01606-16. [Google Scholar] [CrossRef]
- Rodriguez, W.; Srivastav, K.; Muller, M. C19ORF66 Broadly Escapes Virus-Induced Endonuclease Cleavage and Restricts Kaposi’s Sarcoma-Associated Herpesvirus. J. Virol. 2019, 93, e00373-19. [Google Scholar] [CrossRef]
- Wang, X.; Xuan, Y.; Han, Y.; Ding, X.; Ye, K.; Yang, F.; Gao, P.; Goff, S.P.; Gao, G. Regulation of HIV-1 Gag-Pol Expression by Shiftless, an Inhibitor of Programmed -1 Ribosomal Frameshifting. Cell 2019, 176, 625–635.e614. [Google Scholar] [CrossRef]
- Kinast, V.; Plociennikowska, A.; Bracht, T.A.; Todt, D.; Brown, R.J.P.; Boldanova, T.; Zhang, Y.; Bruggemann, Y.; Friesland, M.; Engelmann, M.; et al. C19orf66 is an interferon-induced inhibitor of HCV replication that restricts formation of the viral replication organelle. J. Hepatol. 2020, 73, 549–558. [Google Scholar] [CrossRef]
- Wu, Y.; Yang, X.; Yao, Z.; Dong, X.; Zhang, D.; Hu, Y.; Zhang, S.; Lin, J.; Chen, J.; An, S.; et al. C19orf66 interrupts Zika virus replication by inducing lysosomal degradation of viral NS3. PLoS Negl. Trop. Dis. 2020, 14, e0008083. [Google Scholar] [CrossRef]
- Hanners, N.W.; Mar, K.B.; Boys, I.N.; Eitson, J.L.; De la Cruz-Rivera, P.C.; Richardson, R.B.; Fan, W.C.; Wight-Carter, M.; Schoggins, J.W. Shiftless inhibits flavivirus replication in vitro and is neuroprotective in a mouse model of Zika virus pathogenesis. Proc. Natl. Acad. Sci. USA 2021, 118, e2111266118. [Google Scholar] [CrossRef]
- Napthine, S.; Hill, C.H.; Nugent, H.C.M.; Brierley, I. Modulation of Viral Programmed Ribosomal Frameshifting and Stop Codon Readthrough by the Host Restriction Factor Shiftless. Viruses 2021, 13, 1230. [Google Scholar] [CrossRef]
- Wang, H.; Kong, N.; Jiao, Y.; Dong, S.; Sun, D.; Chen, X.; Zheng, H.; Tong, W.; Yu, H.; Yu, L.; et al. EGR1 Suppresses Porcine Epidemic Diarrhea Virus Replication by Regulating IRAV To Degrade Viral Nucleocapsid Protein. J. Virol. 2021, 95, e0064521. [Google Scholar] [CrossRef]
- Yu, D.; Zhao, Y.; Pan, J.; Yang, X.; Liang, Z.; Xie, S.; Cao, R. C19orf66 Inhibits Japanese Encephalitis Virus Replication by Targeting -1 PRF and the NS3 Protein. Virol. Sin. 2021, 36, 1443–1455. [Google Scholar] [CrossRef]
- Rodriguez, W.; Mehrmann, T.; Muller, M. Shiftless Restricts Viral Gene Expression and Influences RNA Granule Formation during KSHV lytic replication. bioRxiv 2022. [Google Scholar] [CrossRef]
- Wu, X.; Thi, V.L.D.; Huang, Y.; Billerbeck, E.; Saha, D.; Hoffmann, H.-H.; Wang, Y.; Silva, L.A.V.; Sarbanes, S.; Sun, T. Intrinsic immunity shapes viral resistance of stem cells. Cell 2018, 172, 423–438.e25. [Google Scholar] [CrossRef] [PubMed]
- Green, J.L.; Osterhout, R.E.; Klova, A.L.; Merkwirth, C.; McDonnell, S.R.; Zavareh, R.B.; Fuchs, B.C.; Kamal, A.; Jakobsen, J.S. Molecular characterization of type I IFN-induced cytotoxicity in bladder cancer cells reveals biomarkers of resistance. Mol. Ther. Oncol. 2021, 23, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Aso, H.; Ito, J.; Koyanagi, Y.; Sato, K. Comparative description of the expression profile of interferon-stimulated genes in multiple cell lineages targeted by HIV-1 infection. Front. Microbiol. 2019, 10, 429. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Jung, K.; Saif, L.J.; Wang, Q. Porcine epidemic diarrhea virus (PEDV): An update on etiology, transmission, pathogenesis, and prevention and control. Virus Res. 2020, 286, 198045. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Boo, S.H.; Kim, Y.K. The emerging role of RNA modifications in the regulation of mRNA stability. Exp. Mol. Med. 2020, 52, 400–408. [Google Scholar] [CrossRef]
- Gillen, S.L.; Giacomelli, C.; Hodge, K.; Zanivan, S.; Bushell, M.; Wilczynska, A. Differential regulation of mRNA fate by the human Ccr4-Not complex is driven by coding sequence composition and mRNA localization. Genome Biol. 2021, 22, 284. [Google Scholar] [CrossRef]
- Turner, M.; Diaz-Munoz, M.D. RNA-binding proteins control gene expression and cell fate in the immune system. Nat. Immunol. 2018, 19, 120–129. [Google Scholar] [CrossRef]
- Dremel, S.E.; Sivrich, F.L.; Tucker, J.M.; Glaunsinger, B.A.; DeLuca, N.A. Manipulation of RNA polymerase III by herpes simplex virus-1. Nat. Commun. 2022, 13, 623. [Google Scholar] [CrossRef] [PubMed]
- Gilbertson, S.; Federspiel, J.D.; Hartenian, E.; Cristea, I.M.; Glaunsinger, B. Changes in mRNA abundance drive shuttling of RNA binding proteins, linking cytoplasmic RNA degradation to transcription. eLife 2018, 7, e37663. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Qin, Q.; Wu, C.; Li, H.; Shou, J.N.; Yang, Y.; Gu, M.; Ma, C.; Lin, W.; Zou, Y. Downregulated NDR1 protein kinase inhibits innate immune response by initiating an miR146a-STAT1 feedback loop. Nat. Commun. 2018, 9, 2789. [Google Scholar] [CrossRef] [PubMed]
- Rigby, R.E.; Wise, H.M.; Smith, N.; Digard, P.; Rehwinkel, J. PA-X antagonises MAVS-dependent accumulation of early type I interferon messenger RNAs during influenza A virus infection. Sci. Rep. 2019, 9, 7216. [Google Scholar] [CrossRef]
- Zhao, Y.; Ye, X.; Shehata, M.; Dunker, W.; Xie, Z.; Karijolich, J. The RNA quality control pathway nonsense-mediated mRNA decay targets cellular and viral RNAs to restrict KSHV. Nat. Commun. 2020, 11, 3345. [Google Scholar] [CrossRef]
- Uno, N.; Ross, T.M. Dengue virus and the host innate immune response. Emerg. Microbes Infect. 2018, 7, 167. [Google Scholar] [CrossRef]
- Wang, K.; Zou, C.; Wang, X.; Huang, C.; Feng, T.; Pan, W.; Wu, Q.; Wang, P.; Dai, J. Interferon-stimulated TRIM69 interrupts dengue virus replication by ubiquitinating viral nonstructural protein 3. PLoS Pathog. 2018, 14, e1007287. [Google Scholar] [CrossRef]
- Singh, P.K.; Singh, S.; Farr, D.; Kumar, A. Interferon-stimulated gene 15 (ISG15) restricts Zika virus replication in primary human corneal epithelial cells. Ocul. Surf. 2019, 17, 551–559. [Google Scholar] [CrossRef]
- Desole, G.; Sinigaglia, A.; Riccetti, S.; Masi, G.; Pacenti, M.; Trevisan, M.; Barzon, L. Modelling neurotropic flavivirus infection in human induced pluripotent stem cell-derived systems. Int. J. Mol. Sci. 2019, 20, 5404. [Google Scholar] [CrossRef]
- Mattijssen, S.; Kozlov, G.; Fonseca, B.D.; Gehring, K.; Maraia, R.J. LARP1 and LARP4: Up close with PABP for mRNA 3′ poly(A) protection and stabilization. RNA Biol. 2021, 18, 259–274. [Google Scholar] [CrossRef]
- Polacek, C.; Friebe, P.; Harris, E. Poly (A)-binding protein binds to the non-polyadenylated 3′ untranslated region of dengue virus and modulates translation efficiency. J. Gen. Virol. 2009, 90, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Zidovec-Lepej, S.; Vilibic-Cavlek, T.; Barbic, L.; Ilic, M.; Savic, V.; Tabain, I.; Ferenc, T.; Grgic, I.; Gorenec, L.; Bogdanic, M. Antiviral cytokine response in neuroinvasive and non-neuroinvasive west nile virus infection. Viruses 2021, 13, 342. [Google Scholar] [CrossRef] [PubMed]
- Khou, C.; Díaz-Salinas, M.A.; da Costa, A.; Préhaud, C.; Jeannin, P.; Afonso, P.V.; Vignuzzi, M.; Lafon, M.; Pardigon, N. Comparative analysis of neuroinvasion by Japanese encephalitis virulent and vaccine viral strains in an in vitro model of human blood-brain barrier. PLoS ONE 2021, 16, e0252595. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, S.; Richner, J.M.; Clyde, K.; Lee, Y.J.; Glaunsinger, B.A. Host Shutoff Is a Conserved Phenotype of Gammaherpesvirus Infection and Is Orchestrated Exclusively from the Cytoplasm. J. Virol. 2009, 83, 9554–9566. [Google Scholar] [CrossRef]
- Covarrubias, S.; Gaglia, M.M.; Kumar, G.R.; Wong, W.; Jackson, A.O.; Glaunsinger, B.A. Coordinated destruction of cellular messages in translation complexes by the gammaherpesvirus host shutoff factor and the Mammalian Exonuclease Xrn1. PLoS Pathog. 2011, 7, e1002339. [Google Scholar] [CrossRef]
- Richner, J.M.; Clyde, K.; Pezda, A.C.; Cheng, B.Y.H.; Wang, T.; Kumar, G.R.; Covarrubias, S.; Coscoy, L.; Glaunsinger, B. Global mrna degradation during lytic gammaherpesvirus infection contributes to establishment of viral latency. PLoS Pathog. 2011, 7, e1002150. [Google Scholar] [CrossRef]
- He, T.; Wang, M.; Cheng, A.; Yang, Q.; Wu, Y.; Jia, R.; Liu, M.; Zhu, D.; Chen, S.; Zhang, S.; et al. Host shutoff activity of VHS and SOX-like proteins: Role in viral survival and immune evasion. Virol. J. 2020, 17, 68. [Google Scholar] [CrossRef]
- Daly, R.; Khaperskyy, D.A.; Gaglia, M.M. Fine-tuning a blunt tool: Regulation of viral host shutoff RNases. PLoS Pathog. 2020, 16, e1008385. [Google Scholar] [CrossRef]
- Clyde, K.; Glaunsinger, B.A. Deep sequencing reveals direct targets of gammaherpesvirus-induced mRNA decay and suggests that multiple mechanisms govern cellular transcript escape. PLoS ONE 2011, 6, e19655. [Google Scholar] [CrossRef]
- Schubert, K.; Karousis, E.D.; Jomaa, A.; Scaiola, A.; Echeverria, B.; Gurzeler, L.-A.; Leibundgut, M.; Thiel, V.; Mühlemann, O.; Ban, N. SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat. Struct. Mol. Biol. 2020, 27, 959–966. [Google Scholar] [CrossRef]
- Gaucherand, L.; Porter, B.K.; Levene, R.E.; Price, E.L.; Schmaling, S.K.; Rycroft, C.H.; Kevorkian, Y.; McCormick, C.; Khaperskyy, D.A.; Gaglia, M.M. The influenza A virus endoribonuclease PA-X usurps host mRNA processing machinery to limit host gene expression. Cell Rep. 2019, 27, 776–792.e777. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, Y.; Miles, J.; Muller, M. Nonstructural protein 1 (nsp1) widespread RNA decay phenotype varies among Coronaviruses. bioRxiv 2022. [Google Scholar] [CrossRef]
- Muller, M.; Hutin, S.; Marigold, O.; Li, K.H.; Burlingame, A.; Glaunsinger, B.A. A Ribonucleoprotein Complex Protects the Interleukin-6 mRNA from Degradation by Distinct Herpesviral Endonucleases. PLoS Pathog. 2015, 11, e1004899. [Google Scholar] [CrossRef]
- Muller, M.; Glaunsinger, B.A. Nuclease escape elements protect messenger RNA against cleavage by multiple viral endonucleases. PLoS Pathog. 2017, 13, e1006593. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Curnutte, H.A.; Trcek, T. RNA granules: A view from the RNA perspective. Molecules 2020, 25, 3130. [Google Scholar] [CrossRef]
- Tauber, D.; Tauber, G.; Parker, R. Mechanisms and regulation of RNA condensation in RNP granule formation. Trends Biochem. Sci. 2020, 45, 764–778. [Google Scholar] [CrossRef]
- Riggs, C.L.; Kedersha, N.; Ivanov, P.; Anderson, P. Mammalian stress granules and P bodies at a glance. J. Cell Sci. 2020, 133, jcs242487. [Google Scholar] [CrossRef]
- Corbet, G.; Parker, R. RNP Granule Formation: Lessons from P-Bodies and Stress Granules. Cold Spring Harb. Symp. Quant. Biol. 2019, 84, 203–215. [Google Scholar] [CrossRef]
- Zhou, H.; Mangelsdorf, M.; Liu, J.; Zhu, L.; Wu, J.Y. RNA-binding proteins in neurological diseases. Sci. China Life Sci. 2014, 57, 432–444. [Google Scholar] [CrossRef]
- Kedersha, N.; Anderson, P. Mammalian stress granules and processing bodies. Methods Enzymol. 2007, 431, 61–81. [Google Scholar]
- Min, H.; Xu, L.; Parrott, R.; Overall, C.C.; Lillich, M.; Rabjohns, E.M.; Rampersad, R.R.; Tarrant, T.K.; Meadows, N.; Fernandez-Castaneda, A. Mesenchymal stromal cells reprogram monocytes and macrophages with processing bodies. Stem Cells 2021, 39, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Hutchins, E.J.; Piacentino, M.L.; Bronner, M.E. P-bodies are sites of rapid RNA decay during the neural crest epithelial-mesenchymal transition. bioRxiv 2020. [Google Scholar] [CrossRef]
- Fan, A.C.; Leung, A.K. RNA granules and diseases: A case study of stress granules in ALS and FTLD. In RNA Processing; Springer: Cham, Switzerland, 2016; pp. 263–296. [Google Scholar]
- Anderson, P.; Kedersha, N.; Ivanov, P. Stress granules, P-bodies and cancer. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2015, 1849, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Turakhiya, A.; Meyer, S.R.; Marincola, G.; Böhm, S.; Vanselow, J.T.; Schlosser, A.; Hofmann, K.; Buchberger, A. ZFAND1 recruits p97 and the 26S proteasome to promote the clearance of arsenite-induced stress granules. Mol. Cell 2018, 70, 906–919.e7. [Google Scholar] [CrossRef]
- Mateju, D.; Eichenberger, B.; Voigt, F.; Eglinger, J.; Roth, G.; Chao, J.A. Single-molecule imaging reveals translation of mRNAs localized to stress granules. Cell 2020, 183, 1801–1812.e13. [Google Scholar] [CrossRef]
- Mateju, D.; Chao, J.A. Stress granules: Regulators or by-products? FEBS J. 2022, 289, 363–373. [Google Scholar] [CrossRef]
- Buddika, K.; Huang, Y.-T.; Ariyapala, I.S.; Butrum-Griffith, A.; Norrell, S.A.; O’Connor, A.M.; Patel, V.K.; Rector, S.A.; Slovan, M.; Sokolowski, M. Coordinated repression of pro-differentiation genes via P-bodies and transcription maintains Drosophila intestinal stem cell identity. Curr. Biol. 2022, 32, 386–397.e6. [Google Scholar] [CrossRef]
- Ford, L.; Ling, E.; Kandel, E.R.; Fioriti, L. CPEB3 inhibits translation of mRNA targets by localizing them to P bodies. Proc. Natl. Acad. Sci. USA 2019, 116, 18078–18087. [Google Scholar] [CrossRef]
- Robles-Luna, G.; Furman, N.; Barbarich, M.F.; Carlotto, N.; Attorresi, A.; Garcia, M.L.; Kobayashi, K. Interplay between potato virus X and RNA granules in Nicotiana benthamiana. Virus Res. 2020, 276, 197823. [Google Scholar] [CrossRef]
- Moosa, M.M.; Banerjee, P.R. Subversion of host stress granules by coronaviruses: Potential roles of π-rich disordered domains of viral nucleocapsids. J. Med. Virol. 2020, 92, 2891–2893. [Google Scholar]
- Liu, D.; Ndongwe, T.P.; Puray-Chavez, M.; Casey, M.C.; Izumi, T.; Pathak, V.K.; Tedbury, P.R.; Sarafianos, S.G. Effect of P-body component Mov10 on HCV virus production and infectivity. FASEB J. 2020, 34, 9433–9449. [Google Scholar] [CrossRef]
- Liu, L.; Weiss, E.; Panas, M.D.; Gotte, B.; Sellberg, S.; Thaa, B.; McInerney, G.M. RNA processing bodies are disassembled during Old World alphavirus infection. J. Gen. Virol. 2019, 100, 1375–1389. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.R.; Majerciak, V.; Kruhlak, M.J.; Zheng, Z.M. KSHV inhibits stress granule formation by viral ORF57 blocking PKR activation. PLoS Pathog. 2017, 13, e1006677. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Xu, Z.; Liu, P.; Qin, Y.; Chen, M. Enterovirus 71 2A Protease Inhibits P-Body Formation to Promote Viral RNA Synthesis. J. Virol. 2021, 95, e00922-21. [Google Scholar] [CrossRef] [PubMed]
- Bonenfant, G.; Williams, N.; Netzband, R.; Schwarz, M.C.; Evans, M.J.; Pager, C.T. Zika virus subverts stress granules to promote and restrict viral gene expression. J. Virol. 2019, 93, e00520-19. [Google Scholar] [CrossRef]
- Blázquez, A.-B.; Martín-Acebes, M.A.; Poderoso, T.; Saiz, J.-C. Relevance of oxidative stress in inhibition of eIF2 alpha phosphorylation and stress granules formation during Usutu virus infection. PLoS Negl. Trop. Dis. 2021, 15, e0009072. [Google Scholar] [CrossRef]
- Arakawa, M.; Tabata, K.; Ishida, K.; Kobayashi, M.; Arai, A.; Ishikawa, T.; Suzuki, R.; Takeuchi, H.; Tripathi, L.P.; Mizuguchi, K. Flavivirus recruits the valosin-containing protein (VCP)/NPL4 complex to induce stress granule disassembly for efficient viral genome replication. J. Biol. Chem. 2022, 298, 101597. [Google Scholar] [CrossRef]
- Youn, J.-Y.; Dyakov, B.J.; Zhang, J.; Knight, J.D.; Vernon, R.M.; Forman-Kay, J.D.; Gingras, A.-C. Properties of stress granule and P-body proteomes. Mol. Cell 2019, 76, 286–294. [Google Scholar] [CrossRef]
- Eulalio, A.; Behm-Ansmant, I.; Schweizer, D.; Izaurralde, E. P-body formation is a consequence, not the cause, of RNA-mediated gene silencing. Mol. Cell. Biol. 2007, 27, 3970–3981. [Google Scholar] [CrossRef]
- Campbell, A.M.; De La Cruz Herrera, C.F.; Marcon, E.; Greenblatt, J.; Frappier, L. Epstein-Barr Virus BGLF2 commandeers RISC to interfere with cellular miRNA function. PLoS Pathog. 2022, 18, e1010235. [Google Scholar] [CrossRef]
- Van Treeck, B.; Protter, D.S.; Matheny, T.; Khong, A.; Link, C.D.; Parker, R. RNA self-assembly contributes to stress granule formation and defining the stress granule transcriptome. Proc. Natl. Acad. Sci. USA 2018, 115, 2734–2739. [Google Scholar] [CrossRef]
- Fu, Y.; Zhuang, X. m6A-binding YTHDF proteins promote stress granule formation. Nat. Chem. Biol. 2020, 16, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cohen, T.J. Aggregation of the nucleic acid–binding protein TDP-43 occurs via distinct routes that are coordinated with stress granule formation. J. Biol. Chem. 2019, 294, 3696–3706. [Google Scholar] [CrossRef]
- Park, Y.; Park, J.; Hwang, H.J.; Kim, B.; Jeong, K.; Chang, J.; Lee, J.-B.; Kim, Y.K. Nonsense-mediated mRNA decay factor UPF1 promotes aggresome formation. Nat. Commun. 2020, 11, 3106. [Google Scholar] [CrossRef]
- Pabis, M.; Popowicz, G.M.; Stehle, R.; Fernández-Ramos, D.; Asami, S.; Warner, L.; García-Mauriño, S.M.; Schlundt, A.; Martínez-Chantar, M.L.; Díaz-Moreno, I. HuR biological function involves RRM3-mediated dimerization and RNA binding by all three RRMs. Nucleic Acids Res. 2019, 47, 1011–1029. [Google Scholar] [CrossRef]
- Kenny, P.J.; Kim, M.; Skariah, G.; Nielsen, J.; Lannom, M.C.; Ceman, S. The FMRP–MOV10 complex: A translational regulatory switch modulated by G-Quadruplexes. Nucleic Acids Res. 2020, 48, 862–878. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Liu, X.; Jiang, Y. Roles of MOV10 in Animal RNA Virus Infection. Front. Vet. Sci. 2020, 7, 569737. [Google Scholar]
- Oh, S.-W.; Onomoto, K.; Wakimoto, M.; Onoguchi, K.; Ishidate, F.; Fujiwara, T.; Yoneyama, M.; Kato, H.; Fujita, T. Leader-containing uncapped viral transcript activates RIG-I in antiviral stress granules. PLoS Pathog. 2016, 12, e1005444. [Google Scholar] [CrossRef]
- Deater, M.; Tamhankar, M.; Lloyd, R.E. TDRD3 is an antiviral restriction factor that promotes IFN signaling with G3BP1. PLoS Pathog. 2022, 18, e1010249. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Litscher, G.; Watanabe, N.; Wei, W.; Hashimoto, T.; Iwatsubo, T.; Buchman, V.L.; Shelkovnikova, T.A. ALS-linked cytoplasmic FUS assemblies are compositionally different from physiological stress granules and sequester hnRNPA3, a novel modifier of FUS toxicity. Neurobiol. Dis. 2022, 162, 105585. [Google Scholar] [CrossRef]
- Zimmer, M.M.; Kibe, A.; Rand, U.; Pekarek, L.; Ye, L.; Buck, S.; Smyth, R.P.; Cicin-Sain, L.; Caliskan, N. The short isoform of the host antiviral protein ZAP acts as an inhibitor of SARS-CoV-2 programmed ribosomal frameshifting. Nat. Commun. 2021, 12, 7193. [Google Scholar] [CrossRef]
- Zhou, D.; Jia, F.; Li, Q.; Zhang, L.; Chen, Z.; Zhao, Z.; Cui, M.; Song, Y.; Chen, H.; Cao, S. Japanese encephalitis virus NS1′ protein antagonizes interferon beta production. Virol. Sin. 2018, 33, 515–523. [Google Scholar] [CrossRef]
- Halma, M.T.; Ritchie, D.B.; Cappellano, T.R.; Neupane, K.; Woodside, M.T. Complex dynamics under tension in a high-efficiency frameshift stimulatory structure. Proc. Natl. Acad. Sci. USA 2019, 116, 19500–19505. [Google Scholar] [CrossRef] [PubMed]
- Benner, B.E.; Bruce, J.W.; Kentala, J.R.; Murray, M.; Becker, J.T.; Garcia-Miranda, P.; Ahlquist, P.; Butcher, S.E.; Sherer, N.M. Perturbing HIV-1 Ribosomal Frameshifting Frequency Reveals a cis Preference for Gag-Pol Incorporation into Assembling Virions. J. Virol. 2021, 96, e01349-21. [Google Scholar] [CrossRef]
- Anokhina, V.S.; Miller, B.L. Targeting Ribosomal Frameshifting as an Antiviral Strategy: From HIV-1 to SARS-CoV-2. Acc. Chem. Res. 2021, 54, 3349–3361. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, D.B.; Cappellano, T.R.; Tittle, C.; Rezajooei, N.; Rouleau, L.; Sikkema, W.K.; Woodside, M.T. Conformational dynamics of the frameshift stimulatory structure in HIV-1. RNA 2017, 23, 1376–1384. [Google Scholar] [CrossRef] [PubMed]
- Dulude, D.; Berchiche, Y.A.; Gendron, K.; Brakier-Gingras, L.; Heveker, N. Decreasing the frameshift efficiency translates into an equivalent reduction of the replication of the human immunodeficiency virus type 1. Virology 2006, 345, 127–136. [Google Scholar] [CrossRef][Green Version]
- Brakier-Gingras, L.; Charbonneau, J.; Butcher, S.E. Targeting frameshifting in the human immunodeficiency virus. Expert Opin. Ther. Targets 2012, 16, 249–258. [Google Scholar] [CrossRef]
- Clark, M.B.; Janicke, M.; Gottesbuhren, U.; Kleffmann, T.; Legge, M.; Poole, E.S.; Tate, W.P. Mammalian gene PEG10 expresses two reading frames by high efficiency–1 frameshifting in embryonic-associated tissues. J. Biol. Chem. 2007, 282, 37359–37369. [Google Scholar] [CrossRef]
- Belew, A.T.; Meskauskas, A.; Musalgaonkar, S.; Advani, V.M.; Sulima, S.O.; Kasprzak, W.K.; Shapiro, B.A.; Dinman, J.D. Ribosomal frameshifting in the CCR5 mRNA is regulated by miRNAs and the NMD pathway. Nature 2014, 512, 265–269. [Google Scholar] [CrossRef]
- Chiabudini, M.; Tais, A.; Zhang, Y.; Hayashi, S.; Wölfle, T.; Fitzke, E.; Rospert, S. Release factor eRF3 mediates premature translation termination on polylysine-stalled ribosomes in Saccharomyces cerevisiae. Mol. Cell. Biol. 2014, 34, 4062–4076. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Authors | Viruses Studied | Restriction Strategy | Target | RNA Granule Association | Reference |
|---|---|---|---|---|---|
| Suzuki (2016) | DENV (Serotypes 1–4) * | Viral Translation, Viral RNA Stabillity | Genomic RNA | - | [10] |
| Xiong (2016) | HIV-1 | Viral Gene Expression | p24 | - | [8] |
| Balinsky (2017) | DENV and EMCV | Viral RNA Stabillity | Genomic RNA | Localization to P-bodies | [11] |
| Rodriguez (2019) | KSHV | Viral Gene Expression | Early/Delayed Early Genes | - | [12] |
| Wang (2019) | HIV-1 | (-1) Frameshift | Ratio of Gag-Pol | - | [13] |
| Kinast (2020) | HCV | Viral Replication Compartment | Membranous Web, PI(4)P | Localization to Stress Granules | [14] |
| Wu (2020) | ZIKV | Lysosomal Degradation | NS3 | - | [15] |
| Hanners (2021) | YFV and HCV* | Viral Gene Expression | Genomic RNA | - | [16] |
| Wang (2021) | PEDV | Ubiquitinylation-based Degradation | Nucleocapsid (N) Protein | - | [18] |
| Yu (2021) | JEV | (-1) Frameshift, Lysosomal Degradation | NS1′-NS1 ratio, NS3 | - | [19] |
| Rodriguez (2022) | KSHV | Viral Gene Expression | ORF57 | Restricts P-Body Formation, Stress Granule-like Densities | [20] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez, W.; Muller, M. Shiftless, a Critical Piece of the Innate Immune Response to Viral Infection. Viruses 2022, 14, 1338. https://doi.org/10.3390/v14061338
Rodriguez W, Muller M. Shiftless, a Critical Piece of the Innate Immune Response to Viral Infection. Viruses. 2022; 14(6):1338. https://doi.org/10.3390/v14061338
Chicago/Turabian StyleRodriguez, William, and Mandy Muller. 2022. "Shiftless, a Critical Piece of the Innate Immune Response to Viral Infection" Viruses 14, no. 6: 1338. https://doi.org/10.3390/v14061338
APA StyleRodriguez, W., & Muller, M. (2022). Shiftless, a Critical Piece of the Innate Immune Response to Viral Infection. Viruses, 14(6), 1338. https://doi.org/10.3390/v14061338