
Broadly Applicable, Virus-Free Dual Reporter Assay to Identify Compounds Interfering with Membrane Fusion: Performance for HSV-1 and SARS-CoV-2

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmids
2.2. Cell Culture
2.3. Stable Reporter Cell Lines
2.4. Stable Reporter Fusion Assay (SRFIA) with HSV-1 Fusion Machinery
2.5. Stable Reporter Fusion Assay (SRFIA) with SARS-CoV-2 Fusion Machinery
2.6. Measurement of NLuc Activity
2.7. Cytotoxicity Assay XTT
2.8. Microscopical Determination of Syncytia Formation
2.9. Calculation of Zhang Indices
2.10. Statistics
3. Results
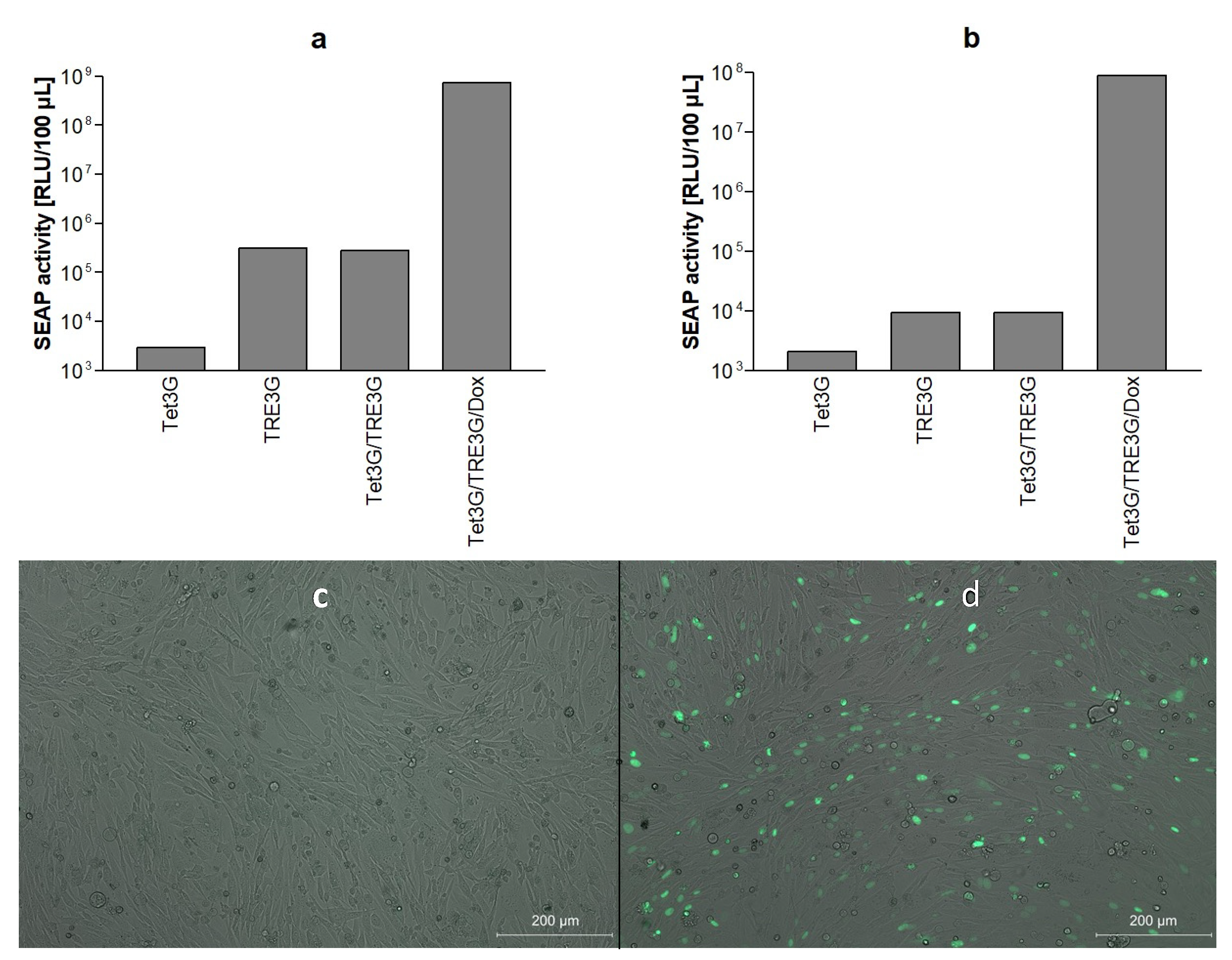
3.1. Bicistronic Reporter Gene Cassette and Stable Vero Reporter Cell Lines
3.2. HSV-1 Fusion Machinery Induces Syncytia in Vero Reporter Cells
3.3. HSV-1 Glycoprotein Mediated Cell–Cell Fusion Results in SEAP Reporter Gene Expression
3.4. Effect of Known Inhibitors of HSV-1 Cell–Cell Fusion on Reporter Gene Expression
3.5. Establishment of Dual secNL/SEAP SRFIA
3.6. Performance of HSV-1-Specific SRFIA as Screening Assay for Fusion Inhibitors on 96-Well Plates
3.7. SARS-CoV-2 S Protein Induces Formation of Syncytia and SEAP Expression in Reporter Cells
3.8. Effect of Umifenovir on Syncytia Formation by the S Protein
3.9. Performance of S Protein-Specific SRFIA as Screening Assay on 96-Well Plates
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harrison, S.C. Viral membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 690–698. [Google Scholar] [CrossRef]
- Harrison, S.C. Viral membrane fusion. Virology 2015, 479–480, 498–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kielian, M.; Rey, F.A. Virus membrane-fusion proteins: More than one way to make a hairpin. Nat. Rev. Microbiol. 2006, 4, 67–76. [Google Scholar] [CrossRef]
- White, J.M.; Delos, S.E.; Brecher, M.; Schornberg, K. Structures and Mechanisms of Viral Membrane Fusion Proteins: Multiple Variations on a Common Theme. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 189–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigant, F.; Santos, N.C.; Lee, B. Broad-spectrum antivirals against viral fusion. Nat. Rev. Microbiol. 2015, 13, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Berkhout, B.; Eggink, D.; Sanders, R.W. Is there a future for antiviral fusion inhibitors? Curr. Opin. Virol. 2012, 2, 50–59. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Seidah, N.G.; Pasquato, A.; Andréo, U. How do enveloped viruses exploit the secretory proprotein convertases to regulate infectivity and spread? Viruses 2021, 13, 1229. [Google Scholar] [CrossRef]
- White, J.M. Viral and cellular membrane fusion proteins. Annu. Rev. Physiol. 1990, 52, 675–697. [Google Scholar] [CrossRef]
- Leroy, H.; Han, M.; Woottum, M.; Bracq, L.; Xie, M.; Benichou, S. Virus-Mediated Cell-Cell Fusion. Int. J. Mol. Sci. 2020, 21, 9644. [Google Scholar] [CrossRef]
- Rajah, M.M.; Bernier, A.; Buchrieser, J.; Schwartz, O. The Mechanism and Consequences of SARS-CoV-2 Spike-Mediated Fusion and Syncytia Formation. J. Mol. Biol. 2021, 434, 167280. [Google Scholar] [CrossRef] [PubMed]
- Okuma, K.; Nakamura, M.; Nakano, S.; Niho, Y.; Matsuura, Y. Host range of human T-cell leukemia virus type I analyzed by a cell fusion-dependent reporter gene activation assay. Virology 1999, 254, 235–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nussbaum, O.; Broder, C.C.; Berger, E.A. Fusogenic mechanisms of enveloped-virus glycoproteins analyzed by a novel recombinant vaccinia virus-based assay quantitating cell fusion-dependent reporter gene activation. J. Virol. 1994, 68, 5411–5422. [Google Scholar] [CrossRef] [Green Version]
- York, J.; Nunberg, J.H. A cell-cell fusion assay to assess arenavirus envelope glycoprotein membrane-fusion activity. Methods Mol. Biol. 2018, 1604, 157–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, N.; Miyauchi, K.; Meng, F.; Iwamoto, A.; Matsuda, Z. Conformational changes of the HIV-1 envelope protein during membrane fusion are inhibited by the replacement of its membrane-spanning domain. J. Biol. Chem. 2010, 285, 14681–14688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atanasiu, D.; Saw, W.T.; Gallagher, J.R.; Hannah, B.P.; Matsuda, Z.; Whitbeck, J.C.; Cohen, G.H.; Eisenberg, R.J. Dual Split Protein-Based Fusion Assay Reveals that Mutations to Herpes Simplex Virus (HSV) Glycoprotein gB Alter the Kinetics of Cell-Cell Fusion Induced by HSV Entry Glycoproteins. J. Virol. 2013, 87, 11332–11345. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Su, P.-Y.; Castro, D.A.; Tripler, T.N.; Hu, Y.; Cook, M.; Ko, A.I.; Farhadian, S.F.; Israelow, B.; Dela Cruz, C.S.; et al. Rapid, reliable, and reproducible cell fusion assay to quantify SARS-CoV-2 spike interaction with hACE2. PLoS Pathog. 2021, 17, e1009683. [Google Scholar] [CrossRef]
- Thakur, N.; Conceicao, C.; Isaacs, A.; Human, S.; Modhiran, N.; McLean, R.K.; Pedrera, M.; Tan, T.K.; Rijal, P.; Townsend, A.; et al. Micro-fusion inhibition tests: Quantifying antibody neutralization of virus-mediated cell-cell fusion. J. Gen. Virol. 2021, 102, 001506. [Google Scholar] [CrossRef]
- Mohamed, F.F.; Anhlan, D.; Schöfbänker, M.; Schreiber, A.; Classen, N.; Hensel, A.; Hempel, G.; Scholz, W.; Kühn, J.; Hrincius, E.R.; et al. Hypericum perforatum and Its Ingredients Hypericin and Pseudohypericin Demonstrate an Antiviral Activity against SARS-CoV-2. Pharmaceuticals 2022, 15, 530. [Google Scholar] [CrossRef]
- Rey, F.A.; Lok, S.M. Common Features of Enveloped Viruses and Implications for Immunogen Design for Next-Generation Vaccines. Cell 2018, 172, 1319–1334. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Buchrieser, J.; Dufloo, J.; Hubert, M.; Monel, B.; Planas, D.; Rajah, M.M.; Planchais, C.; Porrot, F.; Guivel-Benhassine, F.; Van der Werf, S.; et al. Syncytia formation by SARS-CoV-2-infected cells. EMBO J. 2020, 39, e106267. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Evans, J.P.; King, T.; Zheng, Y.-M.; J Whelan, S.P.; Saif, L.; Peeples, M.E.; Liu, S.-L. SARS-CoV-2 Spreads through Cell-to-Cell Transmission. Proc. Natl. Acad. Sci. USA 2021, 119, e2111400119. [Google Scholar] [CrossRef]
- Turner, A.; Bruun, B.; Minson, T.; Browne, H. Glycoproteins gB, gD, and gHgL of Herpes Simplex Virus Type 1 Are Necessary and Sufficient To Mediate Membrane Fusion in a Cos Cell Transfection System. J. Virol. 1998, 72, 873–875. [Google Scholar] [CrossRef] [Green Version]
- Pertel, P.E.; Fridberg, A.; Parish, M.L.; Spear, P.G. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 2001, 279, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Geraghty, R.J.; Krummenacher, C.; Cohen, G.H.; Eisenberg, R.J.; Spear, P.G. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 1998, 280, 1618–1620. [Google Scholar] [CrossRef]
- Montgomery, R.I.; Warner, M.S.; Lum, B.J.; Spear, P.G. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 1996, 87, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Heldwein, E.E.; Lou, H.; Bender, F.C.; Cohen, G.H.; Eisenberg, R.J.; Harrison, S.C. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 2006, 313, 217–220. [Google Scholar] [CrossRef] [Green Version]
- Chowdary, T.K.; Cairns, T.M.; Atanasiu, D.; Cohen, G.H.; Eisenberg, R.J.; Heldwein, E.E. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat. Struct. Mol. Biol. 2010, 17, 882–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laquerre, S.; Argnani, R.; Anderson, D.B.; Zucchini, S.; Manservigi, R.; Glorioso, J.C. Heparan Sulfate Proteoglycan Binding by Herpes Simplex Virus Type 1 Glycoproteins B and C, Which Differ in Their Contributions to Virus Attachment, Penetration, and Cell-to-Cell Spread. J. Virol. 1998, 72, 6119–6130. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.Z.; Person, S.; Warner, S.C.; Zhou, J.H.; DeLuca, N.A. Linker-insertion nonsense and restriction-site deletion mutations of the gB glycoprotein gene of herpes simplex virus type 1. J. Virol. 1987, 61, 714–721. [Google Scholar] [CrossRef] [Green Version]
- Hafezi, W.; Lorentzen, E.U.; Eing, B.R.; Müller, M.; King, N.J.C.; Klupp, B.; Mettenleiter, T.C.; Kühn, J.E. Entry of herpes simplex virus type 1 (HSV-1) into the distal axons of trigeminal neurons favors the onset of nonproductive, silent infection. PLoS Pathog. 2012, 8, e1002679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, D.H.; Marcelletti, J.F.; Khalil, M.H.; Pope, L.E.; Katz, L.R. Antiviral activity of 1-docosanol, an inhibitor of lipid-enveloped viruses including herpes simplex. Proc. Natl. Acad. Sci. USA 1991, 88, 10825–10829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krawczyk, A.; Krauss, J.; Eis-Hubinger, A.M.; Daumer, M.P.; Schwarzenbacher, R.; Dittmer, U.; Schneweis, K.E.; Jager, D.; Roggendorf, M.; Arndt, M.A.E. Impact of Valency of a Glycoprotein B-Specific Monoclonal Antibody on Neutralization of Herpes Simplex Virus. J. Virol. 2011, 85, 1793–1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.H.; Chung, T.D.Y.; Oldenburg, K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef]
- Pope, L.E.; Marcelletti, J.F.; Katz, L.R.; Katz, D.H. Anti-herpes simplex virus activity of n-docosanol correlates with intracellular metabolic conversion of the drug. J. Lipid Res. 1996, 37, 2167–2178. [Google Scholar] [CrossRef]
- Wang, X.; Cao, R.; Zhang, H.; Liu, J.; Xu, M.; Hu, H.; Li, Y.; Zhao, L.; Li, W.; Sun, X.; et al. The anti-influenza virus drug, arbidol is an efficient inhibitor of SARS-CoV-2 in vitro. Cell Discov. 2020, 6, 28. [Google Scholar] [CrossRef]
- Gossen, M.; Bujard, H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. USA. 1992, 89, 5547–5551. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Vink, M.; Klaver, B.; Berkhout, B.; Das, A.T. Optimization of the Tet-On system for regulated gene expression through viral evolution. Gene Ther. 2006, 13, 1382–1390. [Google Scholar] [CrossRef] [PubMed]
- Stieger, K.; Belbellaa, B.; Le Guiner, C.; Moullier, P.; Rolling, F. In vivo gene regulation using tetracycline-regulatable systems. Adv. Drug Deliv. Rev. 2009, 61, 527–541. [Google Scholar] [CrossRef] [PubMed]
- Tristán-Manzano, M.; Justicia-Lirio, P.; Maldonado-Pérez, N.; Cortijo-Gutiérrez, M.; Benabdellah, K.; Martin, F. Externally-Controlled Systems for Immunotherapy: From Bench to Bedside. Front. Immunol. 2020, 11, 2044. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Greenwald, E.; Ahmad, S.; Hur, S. An origin of the immunogenicity of in vitro transcribed RNA. Nucleic Acids Res. 2018, 46, 5239–5249. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.-T.; Sinai, P.; Kitts, P.A.; Kain, S.R. Quantification of Gene Expression with a Secreted Alkaline Phosphatase Reporter System. Biotechniques 1997, 23, 1110–1114. [Google Scholar] [CrossRef]
- Berger, J.; Hauber, J.; Hauber, R.; Geiger, R.; Cullen, B.R. Secreted placental alkaline phosphatase: A powerful new quantitative indicator of gene expression in eukaryotic cells. Gene 1988, 66, 1–10. [Google Scholar] [CrossRef]
- Naoki, O.; Arihiro, K.; Toshiyuki, Y.; Noriko, H.; Fumio, K.; Suyoshi, S.; Makoto, K.; Kentaro, H.; Hattori, M. The genome landscape of the African Green Monkey kidney-derived vero cell line. DNA Res. 2014, 21, 673–683. [Google Scholar] [CrossRef] [Green Version]
- Barrett, P.N.; Mundt, W.; Kistner, O.; Howard, M.K. Vero cell platform in vaccine production: Moving towards cell culture-based viral vaccines. Expert Rev. Vaccines 2009, 8, 607–618. [Google Scholar] [CrossRef]
- Bär, S.; Takada, A.; Kawaoka, Y.; Alizon, M. Detection of Cell-Cell Fusion Mediated by Ebola Virus Glycoproteins. J. Virol. 2006, 80, 2815–2822. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.T.Y.; Spear, P.G. Viral and cellular factors that influence cell fusion induced by herpes simplex virus. Virology 1980, 107, 402–414. [Google Scholar] [CrossRef]
- Leung, D.T.; Sacks, S.L. Docosanol: A topical antiviral for herpes labialis. Expert Opin. Pharmacother. 2004, 5, 2567–2571. [Google Scholar] [CrossRef] [PubMed]
- Blaising, J.; Polyak, S.J.; Pécheur, E.I. Arbidol as a broad-spectrum antiviral: An update. Antivir. Res. 2014, 107, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.P.; Unch, J.; Binkowski, B.F.; Valley, M.P.; Butler, B.L.; Wood, M.G.; Otto, P.; Zimmerman, K.; Vidugiris, G.; MacHleidt, T.; et al. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem. Biol. 2012, 7, 1848–1857. [Google Scholar] [CrossRef] [PubMed]
- England, C.G.; Ehlerding, E.B.; Cai, W. NanoLuc: A Small Luciferase Is Brightening Up the Field of Bioluminescence. Bioconjug. Chem. 2016, 27, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Kawasaki, K.; Ogawa, Y.; Yoshida, Y.; Ohgiya, S.; Ohmiya, Y. Preparation of biotinylated Cypridina luciferase and its use in bioluminescent enzyme immunoassay. Anal. Chem. 2007, 79, 1634–1638. [Google Scholar] [CrossRef]
- Wu, C.; Suzuki-Ogoh, C.; Ohmiya, Y. Dual-reporter assay using two secreted luciferase genes. Biotechniques 2007, 42, 290–292. [Google Scholar] [CrossRef] [Green Version]
- Weyermann, J.; Lochmann, D.; Zimmer, A. A practical note on the use of cytotoxicity assays. Int. J. Pharm. 2005, 288, 369–376. [Google Scholar] [CrossRef]
- Adan, A.; Kiraz, Y.; Baran, Y. Cell Proliferation and Cytotoxicity Assays. Curr. Pharm. Biotechnol. 2016, 17, 1213–1221. [Google Scholar] [CrossRef]
- Hoffmann, M.; Krüger, N.; Schulz, S.; Cossmann, A.; Rocha, C.; Kempf, A.; Nehlmeier, I.; Graichen, L.; Moldenhauer, A.S.; Winkler, M.S.; et al. The Omicron variant is highly resistant against antibody-mediated neutralization: Implications for control of the COVID-19 pandemic. Cell 2022, 185, 447–456.e11. [Google Scholar] [CrossRef]
- Ishikawa, H.; Meng, F.; Kondo, N.; Iwamoto, A.; Matsuda, Z. Generation of a dual-functional split-reporter protein for monitoring membrane fusion using self-associating split GFP. Protein Eng. Des. Sel. 2012, 25, 813–820. [Google Scholar] [CrossRef] [Green Version]
- Saw, W.T.; Matsuda, Z.; Eisenberg, R.J.; Cohen, G.H.; Atanasiu, D. Using a split luciferase assay (SLA) to measure the kinetics of cell-cell fusion mediated by herpes simplex virus glycoproteins. Methods 2015, 90, 68–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martens, S.; McMahon, H.T. Mechanisms of membrane fusion: Disparate players and common principles. Nat. Rev. Mol. Cell Biol. 2008, 9, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, S. Targeting cell signalling pathways to fight the flu: Towards a paradigm change in anti-influenza therapy. J. Antimicrob. Chemother. 2009, 64, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Zhang Index |
|---|---|
| untreated vs. negative control | 0.89 |
| untreated vs. anti-gB 1:100 | 0.65 |
| untreated vs. anti-gB 1:200 | 0.72 |
| untreated vs. anti-gB 1:400 | 0.74 |
| Sample | Zhang Index |
|---|---|
| untreated vs. mock control | 0.94 |
| untreated vs. umifenovir 50 µM | 0.87 |
| untreated vs. umifenovir 20 µM | 0.78 |
| untreated vs. umifenovir 10 µM | 0.66 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Classen, N.; Ulrich, D.; Hofemeier, A.; Hennies, M.T.; Hafezi, W.; Pettke, A.; Romberg, M.-L.; Lorentzen, E.U.; Hensel, A.; Kühn, J.E. Broadly Applicable, Virus-Free Dual Reporter Assay to Identify Compounds Interfering with Membrane Fusion: Performance for HSV-1 and SARS-CoV-2. Viruses 2022, 14, 1354. https://doi.org/10.3390/v14071354
Classen N, Ulrich D, Hofemeier A, Hennies MT, Hafezi W, Pettke A, Romberg M-L, Lorentzen EU, Hensel A, Kühn JE. Broadly Applicable, Virus-Free Dual Reporter Assay to Identify Compounds Interfering with Membrane Fusion: Performance for HSV-1 and SARS-CoV-2. Viruses. 2022; 14(7):1354. https://doi.org/10.3390/v14071354
Chicago/Turabian StyleClassen, Nica, Diana Ulrich, Arne Hofemeier, Marc Tim Hennies, Wali Hafezi, Aleksandra Pettke, Marie-Luise Romberg, Eva U. Lorentzen, Andreas Hensel, and Joachim E. Kühn. 2022. "Broadly Applicable, Virus-Free Dual Reporter Assay to Identify Compounds Interfering with Membrane Fusion: Performance for HSV-1 and SARS-CoV-2" Viruses 14, no. 7: 1354. https://doi.org/10.3390/v14071354